转录组分析流程
推荐帖子: 【生物信息学】 生物信息学分析目录
1. QC 1. 实验 QC
2. QC 2. 数据 QC
3. QC 3. 过滤
4. QC 4. 比对
5. QC 5. 标准化
6. QC 6. 批次效果
7. Common 1.聚类
12. Advanced 1. 选择性剪接分析(AS 分析)
13. Advanced 2. 轨迹分析
a. 测序技术
b. 生物信息学
c. Seurat 管道
d. 蛋白质组学管道
图 1. 分析指南
1. QC 1. 实验 QC(样品级质量控制)
⑴ 定义:评估组织质量的指标
⑵ RNA-seq方法学
① 步骤 1. 纯化 RNA:DNAse 处理以去除基因组内容
② 步骤 2. 使用 PolyA 选择富集 mRNA
③ 步骤 3. 将 RNA 片段化至文库插入片段大小 (200-400 nt)
④ 步骤 4. 将 RNA 转化为 cDNA
⑤ 步骤 5. 准备用于测序的 cDNA(连接接头、扩增、添加条形码)
⑥ 步骤 6. 对片段的一端或两端进行测序(通常每次读取 50、100 或 150 nt)
⑦ 步骤 7. 映射到基因组
⑶ 类型 1. RIN(RNA 完整性编号)
①背景知识:mRNA占总RNA的比例不到3%。 rRNA占80%以上(真核细胞中主要是28S[2kb]和18S[5kb])。
② 采用 Agilent 2100 生物分析仪测量。
③ RIN算法:利用18S与总RNA的比值、28S与总RNA的比值、18S归一化高度等特征。
④ RIN = 10:完整 RNA。
⑤ RIN = 1:RNA完全降解。
⑥ RIN > 7:通常认为适合 RNA-seq 的质量水平。
⑷ 类型2. DV200:FFPE组织中200nt左右的片段百分比,表明RNA片段化
⑸ 类型 3. Cq 值
⑹ 类型 4. 通过吸光度比进行核酸纯度定量
① 260 nm / 280 nm 比率
○ 纯DNA:~1.8
○ 纯RNA:~2.0
○ 260 nm / 280 nm 比率低表明存在蛋白质或苯酚,它们在 280 nm 处有吸收。
② 260 nm/230 nm 比值
○ 纯 DNA/RNA 分子:~2-2.2
○ 260 nm / 230 nm 比率较低表明存在其他污染物。
⑺ 类型 5. 核酸重量定量
① 使用 miRNeasy Mini Kit (QIAGEN) 或类似方法提取 RNA。
② RNA重量标准:至少250ng。
⑻ 类型 6. RNA 质量评分 (RQS)
⑼ 类型 7. ChIP-seq 实验 QC> ① ChIP 级单克隆抗体:通过 ChIP-seq 进行预测试。所测试的商业生产抗体中有 20-30% 无法满足 ChIP-seq 要求。
② qPCR:最好使用 ChIP 样本测试阳性对照区域。希望检测到比 IgG(非特异性抗体)富集 >10-12 倍的结果。
③ 生物素化转录因子:允许链霉亲和素上的因子下拉。独立于抗体。
④ CUT&RUN、CUT&Tag 和 ChIP-exo 是提高 ChIP-seq 中峰的分辨率和信噪比的方法。
⑽ 类型 8. ATAC-seq 实验 QC
① Tn5 浓度:相对于 DNA 浓度,较高的 Tn5 浓度会增加启动子和增强子处的 ATAC-seq 信号强度,同时减小片段大小。
② 测序通道簇密度:改变片段长度分布和 TSS 富集。
⑾ 类型 9. 空间转录组学实验 QC(参考)
表 1. 空间转录组学实验 QC
①新鲜冰冻组织比FFPE具有更高的RNA完整性,但组织形态质量较差。
② 对于 FFPE 样品,DV200 是更合适的指标。
③ 与基于斑点的 ST 方法相比,Xenium 和 CosMx 等基于图像的空间转录组学平台可以更好地耐受 RNA 降解。
2. QC 2. 数据 QC(序列级质量控制)
⑴ 定义:评估数据质量的指标
⑵ 类型1:数据本身的QC指标:用于通过人工检查或与其他数据集比较来外部有效性确认
① QC 指标
○ dUTP方法:“_1.fastq”代表第一链(反义),“_2.fastq”代表第二条链(有义;原始RNA序列)
○ 基础质量
○ 映射率
○ 可映射性过滤器
○ 类型 1. 唯一性:每个序列从特定碱基开始且具有特定长度的唯一性
○ 类型 2. 对齐性:k-mer 序列如何独特地与基因组区域对齐(最多允许 2 个不匹配)
○ 可映射性分数:S = 1 / 在基因组中找到的匹配数
○ 长读长可以解决高度相似区域之间的映射问题:无论读长如何,基因组的某些区域都很麻烦。
○ 非编码RNA比例
○ 非编码RNA比例高、重复率高表明RNA质量差
○ GC含量
○ GC 含量高:表明存在潜在的 rRNA 污染;需要过滤5S、18S、28S rRNA
○ GC 含量低:表明逆转录存在潜在问题
○ 与CpG岛有关。
○ 阅读重复内容
○ PCR 重复项:重复项,仅是 PCR 过程中相同核酸分子的复制。
○ 如果序列完全相同,则视为 PCR 重复。如果它们相似,则它们被视为生物重复。
○ 在双端实验中,重复发生在双端水平。
○ 一般来说,DNA-seq涉及去除重复,而RNA-seq则不然:在RNA-seq中,相同的序列可能会重复出现,不仅是由于技术上的重复,还因为高表达转录本或短基因。删除这些生物重复可以减少数据的动态范围或降低统计功效。
○ 重复率增加的可能性可能会随着 PCR 过程中循环次数的增加而增加:通过检查重复率与 PCR 循环次数之间的相关性,可以区分技术重复和生物重复。»> ○ 删除重复项的工具:Samtools、Picard、Trimmomatic、Trim Galore!、fastp
○ 独特分子(UMI)
○ 如果独特分子计数低于 10%,则 RNA 质量较低
○ 测序深度
○ 读取次数
○ 如果是选择性剪接或等位基因特异性表达:建议 >5000 万次读取
○ 核糖耗尽文库的 DEG 分析:建议总读数约为 50-6000 万次
○ 与 Poly-A 选择的文库相比,建议去核糖文库的测序深度是 Poly-A 选择的文库的两倍,因为去核糖的文库可以捕获 Poly-A 选择的文库无法捕获的更多种类的 RNA(例如,tRNA、rRNA、不成熟的 RNA)。
○ 测序读长
○ 小片段:接头结合开始,需要接头修剪。
图2. 片段较短时读取适配器的原因
○ 与短读长测序相比,长读长测序的优点:每个核苷酸的成本更低、作图更准确、识别剪接点的能力、检测等位基因特异性表达的能力、解析重复序列的能力。
○ 与短读长测序相比,长读长测序的缺点:总体成本较高,每次读取的成本较高,需要更多的接头修剪。长读长测序(PacBio、Oxford Nanopore)由于其测序方法涉及重复读取和单分子测序,因此更可能含有接头序列。
○ 适配器顺序
○ 接头序列:引入人工 DNA 序列,以确保测序的 DNA 片段附着到流动池上。有时,90% 到 20% 的适配器部分仍保持连接状态。
○ 问题 1: 由于接头序列是人工的,它们可能会导致比对和变体调用失败或引入偏差。
○ 问题 2: 接头序列包含相同的序列,导致覆盖率分析和差异基因表达 (DEG) 分析出现偏差。
○ 要删除接头序列,可使用 AdaptorRemoval、Cutadapt(基于 Python)、Trimmomatic、bbduk.sh、Trim_Galore 和 BBMap 等工具。
○ 外显子部分
○ 在poly-A(+) RNA-seq中,外显子reads占50%到70%。
○ 在 rRNA(-) RNA-seq 的情况下,外显子读数的比例减少。
○ 双端与单端
○ 单端读取:每个文库片段仅从单端测序。
○ 配对末端读取:每个文库片段从两端测序。
○ 双端 (PE) 读取比单端 (SE) 读取更准确,但价格大约是单端 (SE) 读取的两倍。
○ 如果目标只是计算 DEG 分析的基因计数,SE 就足够了。
○ 当 RNA 显着降解时,建议使用 SE。
○ 最好避免在短片段上使用 PE,以防止因对相同核苷酸进行测序而导致效率低下。
图 3. 双端 (PE) 测序中可能出现的效率低下问题
○ 配对读取
○ 较长的 DNA 片段被环化,并对来自联合区域的读数进行测序(两端)。
○ 链特异性 (ssRNA-seq)
○ 可以正向或反向。
○ Poly-A 选择与 Ribo 耗尽
○ 核糖耗尽文库的优点
○ 即使在 RNA 降解的情况下也能工作:cDNA 片段不均匀且短。 Poly-A 选择高度偏向 3’ 端,因此准确性较低。
○ 适合研究非编码RNA。»> ○ 核糖耗尽文库的缺点
○ 贵。
○ 包含大量无意义的读取。
② ChIP-seq 的 QC 指标
○ 映射比例
○ Read 深度:ENCODE 建议 TF(组蛋白修饰)≥1000 万个唯一映射的 read。
○ 库复杂性
○ 背景均匀性(有偏差)
○ 【GC峰会偏差】(https://www.sciencedirect.com/science/article/pii/S1046202320300591)
○ qPCR 富集
○ 片段大小分布
○ 通过 NanoDrop 输入 DNA 质量
○ 【互相关分析】(https://pmc.ncbi.nlm.nih.gov/articles/PMC3431496/):NSC(归一化链系数)、RSC(相对链相关性)
○ FRiP(峰值中的读数分数)(ref1、ref2)、RUP(峰值下的读数):ChIP-seq 数据集中落入峰值的读数比例。 ENCODE 建议 FRiP (RUP) ≥ 1%。
○ SPOT(标签中的信号部分):表示信噪比良好。
○ denQCi、simQCi、QC-STAMP(参考)
○ 基序分析:包含 TF 基序的峰的百分比是多少?该基序是否倾向于出现在峰的中间?预计不会出现所有峰,因为 TF 可能作为蛋白质复合物或异二聚体的一部分结合。
○ 片段长度分布:指片段长度的分布,如单个核小体、二聚体、三聚体等。与CUT&Tag、ATAC-seq等技术相关。
○ IP 富集强度:表示读数与 bin 比例的比率的图表。可以使用 deepTools 或 CHANCE 等工具进行分析。这利用了峰值比在整个窗口中分布狭窄的事实。
○ 链滞后:实际结合位点信号在 + 链和 - 链上的信号之间表现出滞后。 ZINBA 和 PePR 等工具可去除不表现出链滞后的伪影信号。
③ ATAC-seq 的 QC 指标
○ FastQC:例如,“每个碱基序列内容”可用于评估Tn5转座酶的整合偏倚。» ○ ataqv 包提供了 35 个 QC 指标,如下(ref):片段长度分布,% 是高质量和常染色体的读数,% 读取正确配对的末端映射,与重复的常染色体比对的读取百分比、短单核小体比率、TSS 富集、峰中的重复分数、峰外的重复分数、峰重复比率、峰中高质量常染色体读数的累积分数、峰内基因组的累积分数、作图质量的分布、总读段数、标记为辅助的比对百分比、标记为补充的比对百分比、标记为重复的比对百分比、平均作图质量、中位作图质量,未映射的读段百分比,具有未映射配偶的读段百分比,QC 失败读段百分比,未配对读段百分比,比对质量为 0 的读段百分比,配对和作图但处于 RF 方向的读段百分比,配对和作图但处于 FF 方向的读段百分比,配对和作图但处于 RR 方向的读段百分比,配对和作图但在不同染色体上的读段百分比,配对和作图但离配偶太远的读段百分比,%配对和映射但不正确的读数、与常染色体对齐的读数百分比、与线粒体对齐的读数百分比、与重复的线粒体对齐的读数百分比、调用的峰数、片段长度分布距离、来自单个常染色体的读数的最大分数
④ 方法1. 其他数据集:10x Genomics、GEO、ZENODO 等。
⑤ 方法 2. FastQC:需要 Fastq 文件作为输入。基于Java。
○ 2-1. FastQC 和 multiQC:最受欢迎
○ 读取的碱基对质量
○ 读取中的接头序列
○ PCR 重复
○ 代表性过高的序列
○ 每个样品的 GC 分布
○ 2-2. QoRT([参考1](https://pubmed.ncbi.nlm.nih.gov/26187896/),[参考2](https://github.com/hartleys/QoRTs)):非常好
○ RNA 降解:读数分布 5’ → 3’
○ 绞合度检查
○ GC 偏差
○ 2-3. RNASeQC:不错
○ 2-4. RSeQC:曾经有重大错误
○ 2-5. 使用 conda Fastqc 命令 (Linux)
○ 2-6. 下载 SRA (Sequence Reads Archive) 工具包并使用 fastqc 命令
○ 下面是生成文件的示例。
受保护_0
○ 示例文件
⑥ 方法 3. Trimmomatic:以 Fastq 文件作为输入。
⑦ 方法4. Cutadapt:基于Python。
⑧ 方法5. FASTX-Toolkit:以Fastq 文件作为输入。
⑨ 方法6. 映射后QC:以SAM 或BAM 文件作为输入。
○ QC 指标
○ % 独特映射的读数
○ % 读取映射到外显子
○ 复杂性,即 x% 的读取计数被 y% 的基因占用
○ 样品之间的一致性
○ 样本交换:将 Y 染色体、Xist、基因型(例如 SNP)与元数据进行匹配。
○ 5-1. Qplot
○ 5-2. Samtools:
⑩ 方法 7. SnakeMake:具有 QC 功能的集成管道
○
Snakefile: 基于 Python 的 Snakemake 脚本。文件名本身是 Snakefile。
受保护_1
○
config.yaml(可选):Snakemake 工作流程的配置(ref)
○
requirements.txt(可选):包依赖项
○ 输入文件
○ 输出文件> ⑪ 方法 8. QuASAR-QC:适用于 Hi-C 数据。
⑫ 【问题排查】(https://jb243.github.io/pages/2337)
⑶ 类型2: 样本之间的比较或再现性指标:用于内部有效性评估
① 目的 1: 通过检查单个样本中两个相关变量的排名来评估样本质量
○ 示例:研究两个已知相似基因的表达水平的比对
○ 示例:检查两个已知相似的基因是否出现在同一簇中
② 用途2:常用于检查样本对之间的对应关系
③ 目的3:考察具有不同数据分布特征的两个变量之间的相关性
○ 与 QC 分析有些不同的概念
○ 示例:scRNA-seq 中基因 A 表达与 ST(空间转录组学)中 A 表达的相关系数
④ 方法1. 皮尔逊相关系数
○ 定义:对于 X 和 Y 的标准差 σx 和 σy,
○ 特点
○ 在区间或比率尺度上测量的两个变量之间的相关性
○ 关注连续变量
○ 正态性假设
○ 在大多数情况下广泛使用
○ [RStudio]中的计算(http://www.sthda.com/english/wiki/correlation-test- Between-two-variables-in-r#:~:text=Compute%20correlation%20in%20R-,R%20functions,-Correlation%20coefficient%20can)
○
cor(x, y)
○
cor(x, y, method = "pearson")
○
cor.test(x, y)
○
cor.test(x, y, method = "pearson")
⑤ 方法2. Spearman相关系数
○ 定义:定义为 x’=rank(x) 和 y’=rank(x)
○ 特点
○ 在序数尺度上测量两个变量之间相关性的方法
○ 序数变量的非参数方法
○ 对于有很多零的数据有利
○ 对数据偏差或错误敏感
○ 与 Kendall 相关系数相比,产生更高的值
○ 有用的公式
○ [RStudio]中的计算(http://www.sthda.com/english/wiki/correlation-test- Between-two-variables-in-r#:~:text=Compute%20correlation%20in%20R-,R%20functions,-Correlation%20coefficient%20can)
○
cor(x, y, method = "spearman")
○
cor.test(x, y, method = "spearman")
⑥ 方法3. Kendall相关系数
○ 定义:使用一致和不一致对定义
○ 特点
○ 在序数尺度上测量两个变量之间相关性的方法
○ 序数变量的非参数方法
○ 适用于有多个零的数据
○ 对于小样本量或数据中存在许多并列排名时很有用
○ 程序
○ 第 1 步:根据 x 值对 y 值进行升序排序。将每个 y 值表示为 yi。
○ 步骤 2:对于每个 yi,计算 yj > yi(其中 j > i)的一致对的数量。
○ 第 3 步:对于每个 yi,计算 yj < yi(其中 j > i)的不一致对的数量。
○ 步骤 4:定义相关系数。
○ nc: 一致对总数
○ nd: 不一致对的总数
○ n: x 和 y 的大小» ○ [RStudio]中的计算(http://www.sthda.com/english/wiki/correlation-test- Between-two-variables-in-r#:~:text=Compute%20correlation%20in%20R-,R%20functions,-Correlation%20coefficient%20can)
○
cor(x, y, method = "kendall")
○
cor.test(x, y, method = "kendall")
⑦ 方法 4. 经验 CDF(累积分布函数)之间的 Q-Q 图
⑧ 方法 5. 有序 p 值之间的 Q-Q 图
⑨ 对于 Hi-C 测序,可用的方法包括 HiCRep、GenomeDISCO、HiC-Spector 和 QuASAR-Rep。
3。 QC 3. 过滤
⑴ 类型1:数据处理过程中的过滤
① 去除测序数据中不适当的reads,生成修剪后的数据
② 示例 1: 适配器序列
○ 引入人工 DNA 序列,以确保测序的 DNA 片段附着在流动池上。有时,90% 到 20% 的适配器部分仍保持连接状态。
○ 问题 1: 由于接头序列是人工的,它们可能会导致比对和变体调用失败或引入偏差。
○ 问题2: 接头序列包含相同的序列,导致覆盖率分析和差异基因表达(DEG)分析出现偏差。
○ 如果片段太短,则开始读取接头序列。 → 在这种情况下,将进行适配器修整。
○ 长读长测序需要更多的接头修剪。 → 长读长测序(PacBio、Oxford Nanopore)由于其测序方法涉及重复读取和单分子测序,因此更可能含有接头序列。
○ 要删除接头序列,可使用 AdaptorRemoval、Cutadapt(基于 Python)、Trimmomatic、bbduk.sh、Trim_Galore 和 BBMap 等工具。
○ 适配器修剪方法:对齐每个配对端读数并去除不同的部分。
图4. 适配器修剪方法
③ 示例2. 去除低质量碱基:Trimmomatic、Trim_Galore、BBMAP
④ 示例 3. 删除非常短的读数:Trimmomatic、Trim_Galore、SolexQA
⑵ 类型2:下游分析中的过滤
① 2-1. 基因过滤:排除表达量低的基因,避免捕获无信息或不切实际的DEG基因
② 2-2. 条码过滤
○ 情况 1: RNA 表达低(例如,QC 结果不佳)
○ 情况 2: 高 RNA 表达(例如,由于 RNA 扩散造成的污染)
○ 案例 3. Doublet 去除:scdDblFinder(基于 R)、scds 的混合分数(基于 R)、scran 的 doubletCells(基于 R)、DoubletFinder
③ 可以使用R Seurat pipeline中的subset函数、Python scanpy中的矩阵运算等实现。
4。 QC 4. 对齐方式
⑴ 定义:寻找基因组或转录组中特定测序读段位置的过程
⑵ 步骤1:原始数据准备:文件扩展名为.Fastq
① 类型 1: 单端测序 (SES):使用适配器的一侧进行测序
② 类型 2: 双端测序 (PES):使用接头两侧进行测序
○ dUTP方法标准:“_1.fastq”代表第一链(反义),“_2.fastq”代表第二条链(有义;原始RNA序列)
○ 首先通过一个adapter进行测序(获得Read1),然后通过相反的adapter进行测序(获得Read2)。
○ 来自同一 DNA 片段的 Read1 和 Read2 可以轻松匹配,因为它们来自同一簇。» ○ 优点:准确度更高(由于Read1和Read2之间的比较),容易提取DNA突变,容易分析重复序列,容易在不同物种之间作图。
○ 缺点:成本较高,比SES需要更多步骤。
③ 长读长测序
| 短读序列 | 长读序列 | |
|---|---|---|
| 发布年份 | 2000 年代初 | 2010 年代中期 |
| 平均阅读长度 | 150-300 bp | 5,000-10,000 bp |
| 准确度 | 99.9% | 95-99% |
表2. 短读seq和长读seq的比较
图5. 短读seq和长读seq之间的区别
○ 不存在测序间隙,可进行以下分析:
○ 优点1: 选择性剪接分析:可以识别选择性剪接事件和亚型。代表性工具包括纯序列模型,包括 spliceAI、Pangolin、MMSplice。
○ 优势2: 【拷贝数变异(CNV)分析】(https://jb243.github.io/pages/2050#13-advanced-2-trajectory-analysis):例如,重复序列的数量对于亨廷顿病至关重要。
○ 优势3: 促进表观遗传学和转录组学的整合。
○ 类型 1: Pacific Biosciences SMRT(单分子实时)测序:平均读长约为 20 kb。
○ 类型 2: 牛津纳米孔测序:平均读长约为 100 kb。
⑶ 步骤2. 预处理:预处理原始测序数据以提取有价值的汇总信息的过程。
① 流程1. 对齐
图6. 对齐过程
○ 概述
○ 定义:确定两个或多个序列如何相互比对的过程。换句话说,模式匹配。
○ 基序:在给定序列中重复出现的特定模式或子序列。基序通常与生物功能相关。
○ 目的 1. 识别两个序列之间的相似性,以用于映射和组装等过程。
○ 目的2. 变异识别:检测插入、删除和替换等变异。
○ 模式匹配: PFM 和 PWM 的概念
○ 步骤 1. PFM(位置频率矩阵)的构建
○ xij:第 j 个位置出现的核苷酸 i 的计数。
○ 步骤 2. 构造 PWM(位置权重矩阵)
○ PWM 是 PFM 的相对熵或 Kullback-Leibler 散度。
○ pi:伪计数或拉普拉斯估计量(例如 0.25)。
○ qi:观察核苷酸 ( i ) 的预期或背景概率(先验)(例如 0.25)。
○ pi 和 qi 根据信息论确定。
○ 步骤 3. 使用 PWM 为每个 k 聚体分配分数。
» ○ 模式匹配:吉布斯采样
○ 第 1 步: 初始化
○ 1-1. 从 N 个序列中的每一个中随机选择 k-mers,即基序。
○ 1-2. 计算每个位置的A、C、G、T核苷酸的频率。
○ 1-3. 将所有未选为主题的剩余序列视为背景。
○ 1-4. 构建初始 PWM。

图 7. 吉布斯采样初始化
○ 第 2 步: 迭代
○ 2-1. 随机选择N个序列之一。
○ 2-2. 使用除所选序列之外的所有序列构建 PWM。
○ 2-3. 通过考虑所选序列中所有可能的基序来计算分数分布。
○ 2-4. 根据分数分布概率确定新主题。
○ 2-5. 重复步骤2-1至2-4,直至达到最大迭代次数或信息内容不再发生明显变化。
图 8. 吉布斯采样迭代
○ 图案匹配:序列标志
○ 氨基酸或核酸多序列比对的图形表示。
○ 由 Tom Schneider 和 Mike Stephens 开发。
○ y 轴代表信息内容,如信息论中定义。
○ 示例1:当所有核苷酸序列(A、T、G、C)具有相同频率时:最大熵=2。实际熵=2。信息内容=0
○ 实施例2:仅存在一种核苷酸时:最大熵=2。实际熵=0。信息内容=2
○ 示例3:当两个核苷酸频率相同时:最大熵=2。实际熵=1。信息含量=1
○ 模式匹配、压缩: BWT(Burrows-Wheeler 变换)
○ 历史:以大卫·惠勒为中心
○ 第一个获得博士学位计算机科学博士学位:1951 年,剑桥大学。
○ 第一个发明编程语言。
○ 与 Maurice Wilkes 和 Stanley Gill 一起发明了计算机编程中的函数概念。
○ 20 世纪 80 年代初在贝尔实验室担任顾问时发现了 BWT。
○ 十年后与 Michael Burrows 一起出版了 BWT。
○ 步骤1. 当输入字符串为
mississippi时,在末尾追加$,生成所有循环旋转的矩阵。
○ 步骤2. 以
$为第一个字符,按字母顺序对矩阵进行排序,得到BWT矩阵。
○ 下图中,
i称为SA索引,由于特性3,必须将其存储在一起。
图9. 如何计算BWT矩阵
○ 步骤 3. 对 BWT 矩阵的最后一列应用游程编码以获得 BWT 变换。
○ BWT 矩阵的最后一列:
ipssm$pissii
○ 行程编码:
ipssm$piissi或ip2sm$pi2s2i
○ 功能 1. 它倾向于将包含重复的字符串组织成连续的字符。一种高效的压缩技术。
○ 构建 BWT 矩阵时,按字母顺序排序可以让重复序列轻松地按同一字母进行分组。
○ 使用这种方法,即使是包含许多重复序列的人类基因组也可以压缩到大约 750 MB。»> ○ 功能 2. BWT 是可逆的,即给定字符串的 BWT,您可以恢复原始字符串而无需任何附加信息。
○ 如果 BWT 转换为
ippssm$piissi,则 BWT 矩阵的第一列变为$iiiimppssss,这是 BWT 转换按字母顺序排序的序列。
○ 输入字符串的结尾自然是
$,通过查看BWT矩阵的第一行,我们可以确定输入字符串以i$结尾。
○ 在 BWT 矩阵中,以
i$开头的行很容易被识别为第二行,即以i开头的第一行。
○ 按照此方法,我们可以识别 LF(后在先)属性,它允许我们重建原始输入字符串。
○ LF 属性:对于给定字符,BWT 矩阵第一列中的第 k 次出现对应于最后一列中的第 k 次出现。
○ 功能 3. 使用 BWT 进行快速模式匹配:假设模式为
sis。
○ 如果 BWT 转换为
ippssm$piissi,则 BWT 矩阵的第一列变为$iiiimppssss,这是 BWT 转换按字母顺序排序的序列。
○ 要搜索
sis(前两个字母si),请在 BWT 矩阵中查找最后一列为s、第一列为i的行:SA 索引 = 8, 5。
○ 要搜索
sis(最后两个字母is),请在 BWT 矩阵中查找最后一列为i、第一列为s的行:SA 索引 = 6, 3。
○ 方法一. 根据LF属性,
i要通用,对应的SA索引值必须为5和6。
○ 方法 2. SA 索引值必须正好相差 1,这意味着它们应该是 5 和 6。
○ 方法 3. 针对按字母顺序出现在其前面的字母数
count以及该字母的频率occur执行以下算法。如果输出多行,则意味着找到多个匹配项。
受保护_2
○ 由于
sis对应 SA 索引 5,因此mississippi中的匹配模式是从“(5-1) 到 (5-1) + (length-1)”找到的。
○ 类型 1: BLAST、BLAT:分别于 1997 年和 2002 年发布。基于 k-mer 哈希。
○ 类型 2. 批量 RNA 测序比对
○ 2-1. STAR(拼接转录本比对到参考):最常用的比对工具。 ([参考](https://hbctraining.github.io/Intro-to-rnaseq-hpc-O2/lessons/03_alignment.html))。使用后缀数组。速度快,但可能会占用大量内存。
○
--genomeDir:包含基因组索引文件的目录。如果没有文件,请先运行--runMode genomeGenerate。
○
--outFilterMismatchNmax:每对最大不匹配数。
○
--outFilterMultimapNmax:每次读取允许的多重比对数。
○
--runThreadN:线程数。
受保护_3
○ 2-2. HISAT,HISAT2:基于图的 BWT 扩展设计并实现了第一个 GFM(图 FM 索引)。它继承了 TopHat 并执行遗传变异感知比对。
○ HISAT-3N:用于处理核苷酸转换的测序技术,例如亚硫酸氢盐序列中的 C 至 T。
○ 2-3. Bowtie,Bowtie2:使用利用高效压缩技术的 Burrows Wheeler Transform (BWT)。
受保护_4
○ 2-4. Tophat、Tophat2:使用 Burrows Wheeler 变换 (BWT)。»» ○ 假设 70 bp 的内含子长度对于脊椎动物来说是合适的,但对于拟南芥、秀丽隐杆线虫和类似生物体,应使用不同的值。
○ 2-5. BWA(Burrows-Wheeler Aligner):使用 Burrows Wheeler 变换 (BWT)。
○ 2-6. MUMmer
○ 执行 DNA 和蛋白质的快速比对。
○ 原核生物、真菌和哺乳动物基因组的全基因组比对。
○ 30 Mb 真菌基因组向前和向后需要 1~2 分钟。
○ 可以基于6帧平移进行对齐。
○ 使用后缀树进行对齐。
○ 2-7. CLC
○ 2-8. ContextMap2
○ 2-9. CRAC
○ 2-10. GSNAP
○ 2-11. MapSplice2
○ 2-12. Novoalign
○ 2-13. OLego
○ 2-14. 朗姆酒
○ 2-15. SOAPsplice
○ 2-16. 副读
○ 2-17. SOAP、SOAP2:使用 Burrows Wheeler 变换 (BWT)。
○ 2-18. 法斯特先生/夫人
○ 2-19. 伊兰
○ 2-20. 早餐
○ 2-21. BarraCUDA
○ 2-22. 现金x
○ 2-23. 莫西亚克
○ 2-24. 史坦皮
○ 2-25. 虾
○ 2-26. SeqMap
○ 2-27. 滑块
○ 2-28. RMAP
○ 2-29. SSAHA
○ 2-30. bamnostic:与操作系统无关。基于Python。
○ 类型 3. 批量 RNA 测序的伪比对
○ 伪对齐简化了一些对齐步骤,可以更快地处理耗时的过程。
○ 伪对齐比对齐准确度低,但速度更快。
○ 类型 4. scRNA-seq 的比对和计数
○ 4-1. CellRanger:使用 STAR 比对。
○ 类型 5. ST 数据的对齐和计数
○ 5-1. SpaceRanger:仅限 Visium。使用 STAR 对齐但考虑空间条形码。
○ 类型 6. 长读序列比对
○ 曼达洛里恩 (Byrne et al., 2017)
○ 小地图, Minimap2 (李恒, 2018)
○ SQANTI(Tardaguila 等人,2018)
○ 针鱼(ONT,2018)
○ StringTie2(Kovaka 等人,2019)
○ TALON(Wyman 等人,2019)
○ FLAIR(Tang 等人,2020)
○ Bambu(GoekeLab GitHub,2020)
○ 火焰(Tian 等人,2020)
○ 滑石粉(Broseus 等人,2020)
○ LongTron(Wilks 等人,2020)
○ uLTRA(Sahlin 和 Mäkinen,2020)
○ LIQA(胡等人,2021)
○ 2passtools(Parker 等人,2021)
○ MuSTA(Namba 等人,2021)
○ IsoQuant(Prjibelski 等人,2022)
○ 浓缩咖啡(Gao 等人,2023)
○ Bloom2(Nip 等人,2023)
○ 流苏(Kainth 等人,2023)
○ MisER(Liu 等人,2023)
○ 类型 7. 剪接感知对准器(隐式剪接):在比对过程中考虑剪接等 RNA 特异性事件的对准器。
○ 7-1. 巴士凳
○ 7-2. CrypSplice
○ 7-3. IGV
○ 7-4. 床具
○ 7-5. R:伊布
○ 7-6. R:质量
② 流程2. 映射
○ 定义:将序列存储在哈希表中,并使用该表来映射不同序列之间的相似部分。» ○ 背景:所有序列的比较会消耗天文数字的时间,因此有必要为每个序列找到简短的、有代表性的哈希值。
○ 一般流程:
○ 第1步: 将给定序列划分为多个滑动窗口。
○ 第 2 步: 搜索每个滑动窗口内 k 个元素的所有连续序列。
○ 步骤 3: 在 k 个连续序列中,字典顺序最小的序列是该窗口的最小者。
○ 步骤 4: 将四进制数字系统应用于最小值(即 A = 0、C = 1、T = 2、G = 3),将最小值转换为特定值。
○ 步骤5: 将哈希函数应用于最小化器以生成哈希值,旨在缓解碱基分布不对称性并提高数据库效率。
○ 步骤 6: 根据哈希值将每个窗口中的每个最小化器存储在哈希表的相应元素中。
○ 步骤 7: 从查询序列(感兴趣的序列)生成一组最小化器。
○ 步骤 8: 从目标序列(参考序列)生成一组最小化器。
○ 步骤 9: 搜索最小化命中:比较查询和目标序列的最小化集,以探索两个序列之间的相似部分。
○ 关于哈希函数的注意事项:哈希函数不一定需要可逆,但哈希冲突对不应轻易被发现。
○ 映射过程的优点:加快不同序列之间的比较,并确保管道即使在存在噪声或许多 SNP 的情况下也能稳健运行。
○ 类型 1: BLASR
○ 类型 2. DALIGNER
○ 类型 3: MHAP
○ 类型4: GraphMap
○ 类型 5: minimap、minimap2(专为长读长测序设计的映射算法)
③ 过程3:纠错
○ 定义:纠正测序过程中的错误。 Illumina 测序为 0.2% - 0.5% 或长读长测序中更高。
○ 要求:给定序列中的特定 k-mer 频率较低,存在汉明距离较近的替代 k-mer,并且无论该段如何定义,用替代 k-mer 替换它都不会生成任何新定义的 k-mer。在这种情况下,应用替换。
○ 通常包含在装配过程中。
○ 类型 1: pbdagcon
○ 类型 2: falcon_sense
○ 类型 3: nano Correct
④ 流程4. 组装
图10. 组装过程
○ 定义: ○ 定义:将由于技术限制而部分获得的短DNA/RNA片段重建为与参考基因组对齐的图的过程。
○ 1 类. 从头开始组装
○ 需要参考基因组。
○ 需要最少的计算资源。
○ 基因组的完整性和比对仪的性能影响组装的准确性。
○ 两次通过对齐:一种有效检测从头组装中新型剪接点的方法。
○ 步骤 1. 第一遍:将 RNA-seq 读数与参考基因组对齐。»» ○ 步骤 2. 更新剪接点信息:收集在第一遍中检测到的新剪接点并更新参考。
○ 步骤 3. 第二遍:使用更新的剪接点信息重新对齐读数。
○ STAR 两遍对准
○ 单个样本:使用
--twopassMode Basic。
○ 多个样本:正常运行 STAR → 收集每个样本的
SJ.out.tab文件 → 使用--sjdbFileChrStartEnd sj1.tab sj2.tab ...重新运行 STAR。
○ 附加参数 1.
alignIntronMin:最小内含子大小
○ 附加参数 2.
alignSJoverhangMin:识别新剪接点所需的最小核苷酸数(即,为了准确剪接点定位而必须在两个外显子之间对齐的最小核苷酸数)
○ 附加参数 3.
alignSJDBoverhangMin:识别已知剪接点所需的最小核苷酸数(即,为准确剪接点作图必须在两个外显子之间比对的最小核苷酸数)
○ 2 类. 从头组装
○ 不需要参考基因组,消除参考偏差。
○ 通过连接图结构中的重叠读段来构建重叠群;随后在组装后阶段将重叠群与命名参考进行比较。
○ 需要大量计算资源:~1G RAM 用于~1M 76bp 读取。
○ 可以识别参考基因组中不存在的染色体畸变或新亚型。
○ 步骤 1. 重叠群:通过连接具有重叠序列信息的读取构建的连续序列。
图 11. 重叠群
○ E 仅在一端具有同一性,因此仅导致包含 3 个节点。
○ 路径ABCD 有4 个节点,被选为更好的路径。
○ 步骤 2. 创建装配图
○ 2-1. de Bruijn 图
图 12. de Brujin 图
○ 使用 k-mers:构造图,使得每个节点与前一个节点重叠 k-1 个碱基。
○ 当重复序列长于读取长度时就会出现问题:有多种可能的方式来连接重复序列的左侧和右侧。
○ 如果有重复序列,大文库插入片段和配对测序可能会有所帮助。
○ 在 de Bruijn 图中,需要考虑两条链:因此,还必须考虑反向互补。
○ 优点1: 提高了计算序列之间重叠的效率
○ 优点 2: 围绕低 k 聚体计数重复的组装路径直接折叠
○ 优点 3: 修剪终止的低 k 聚体计数序列
○ 2-2. Needleman-Wunsch 图:配对对齐
○ 阶段1. 构建全局对齐图
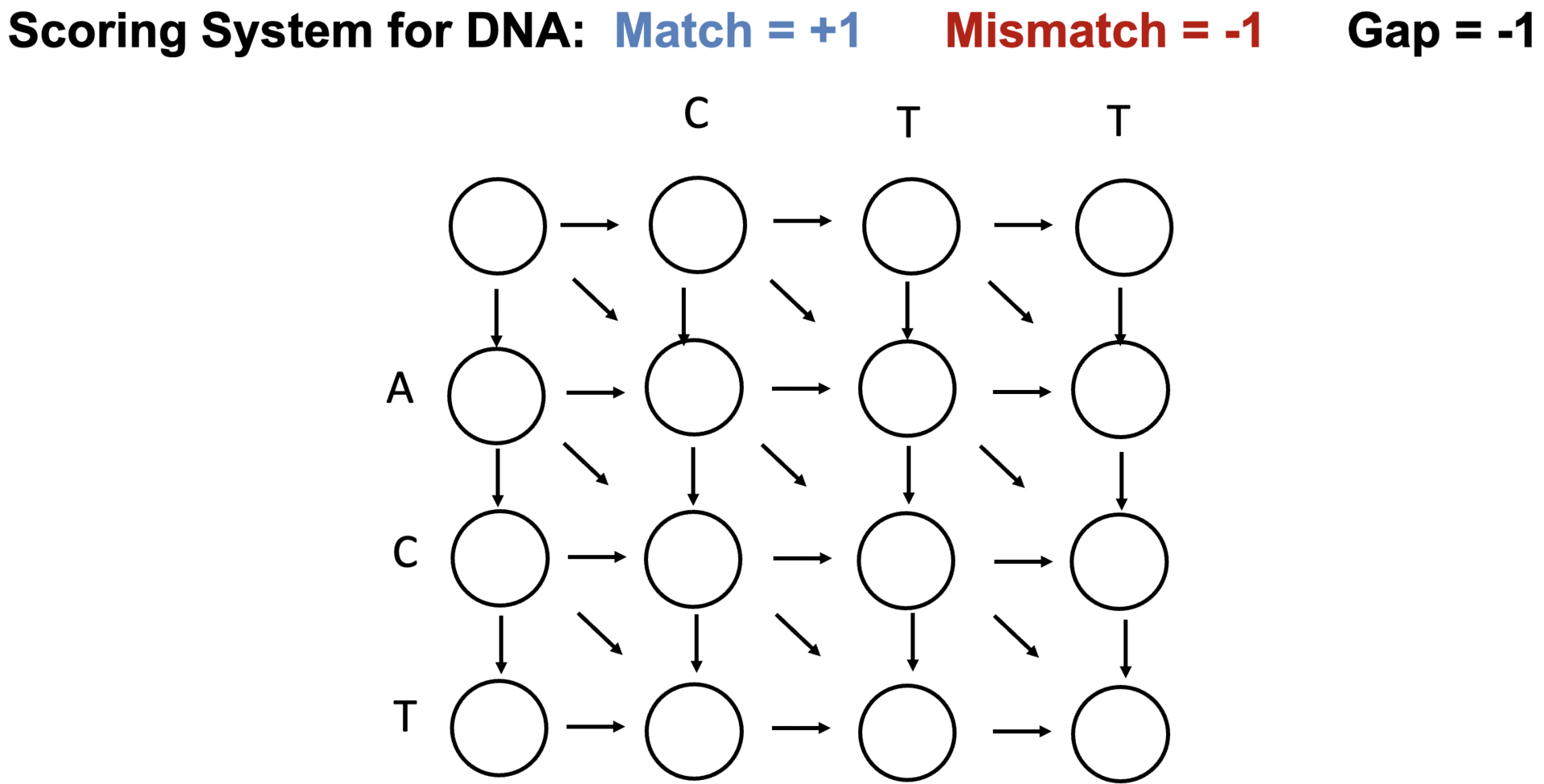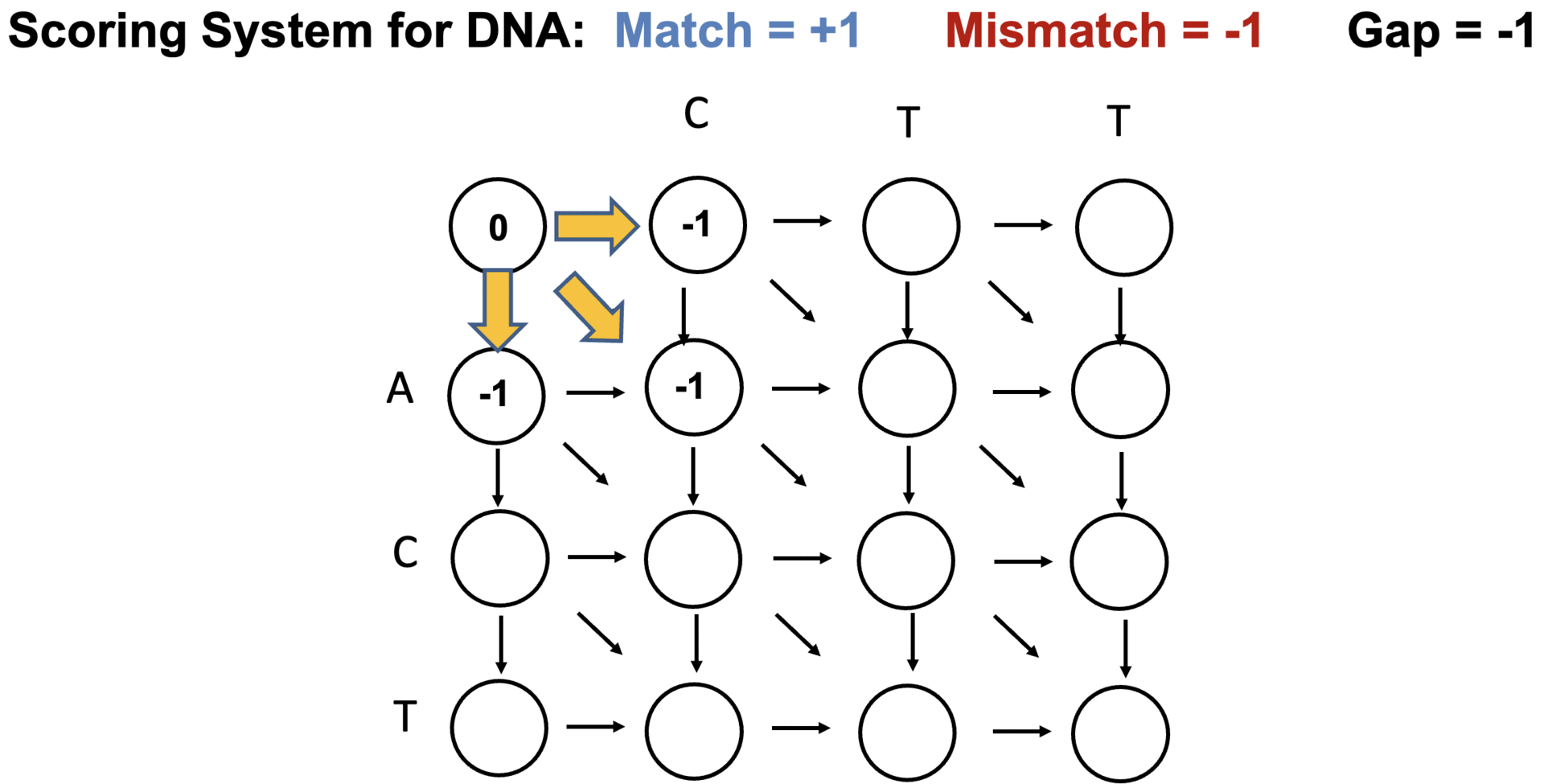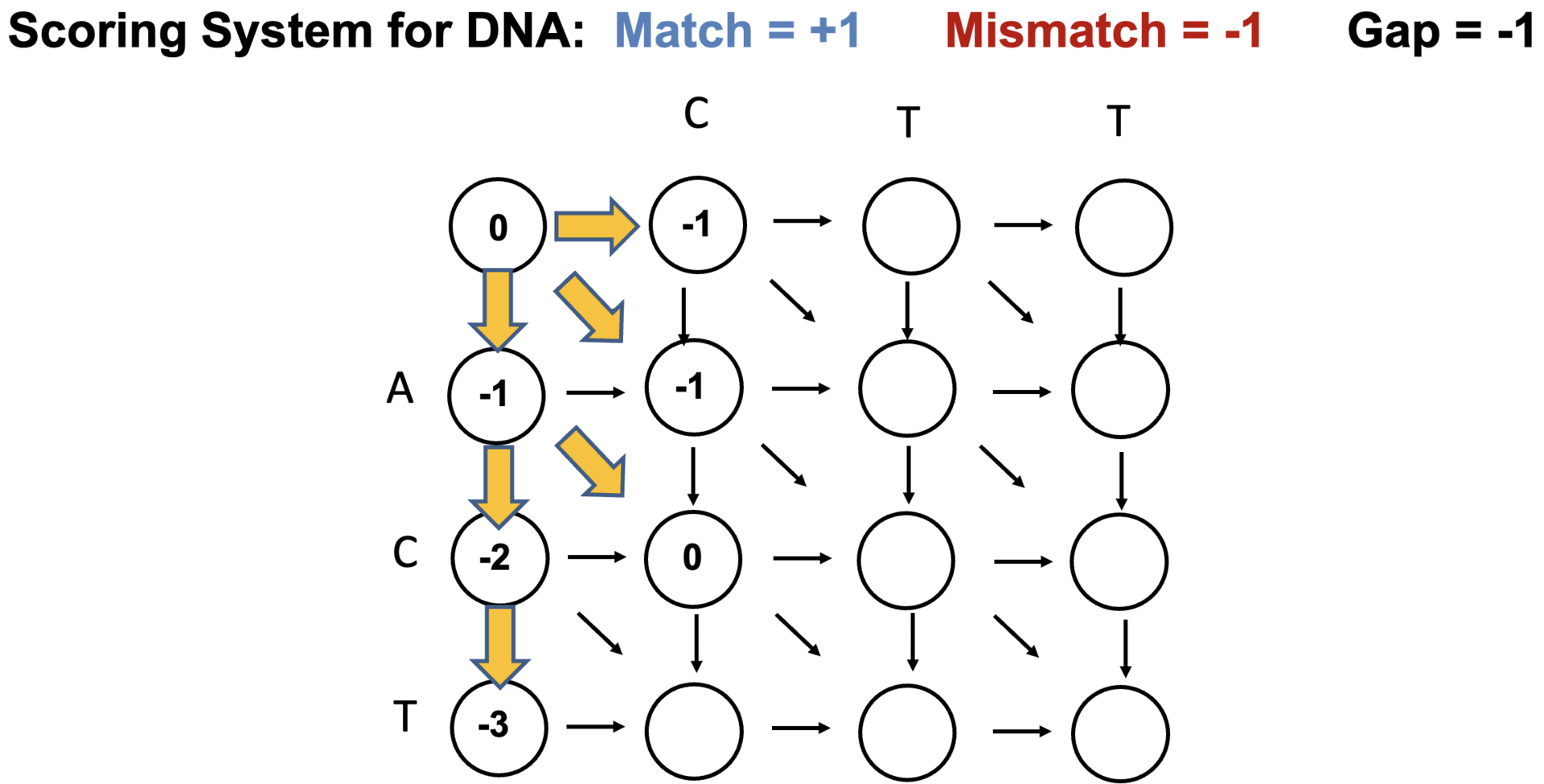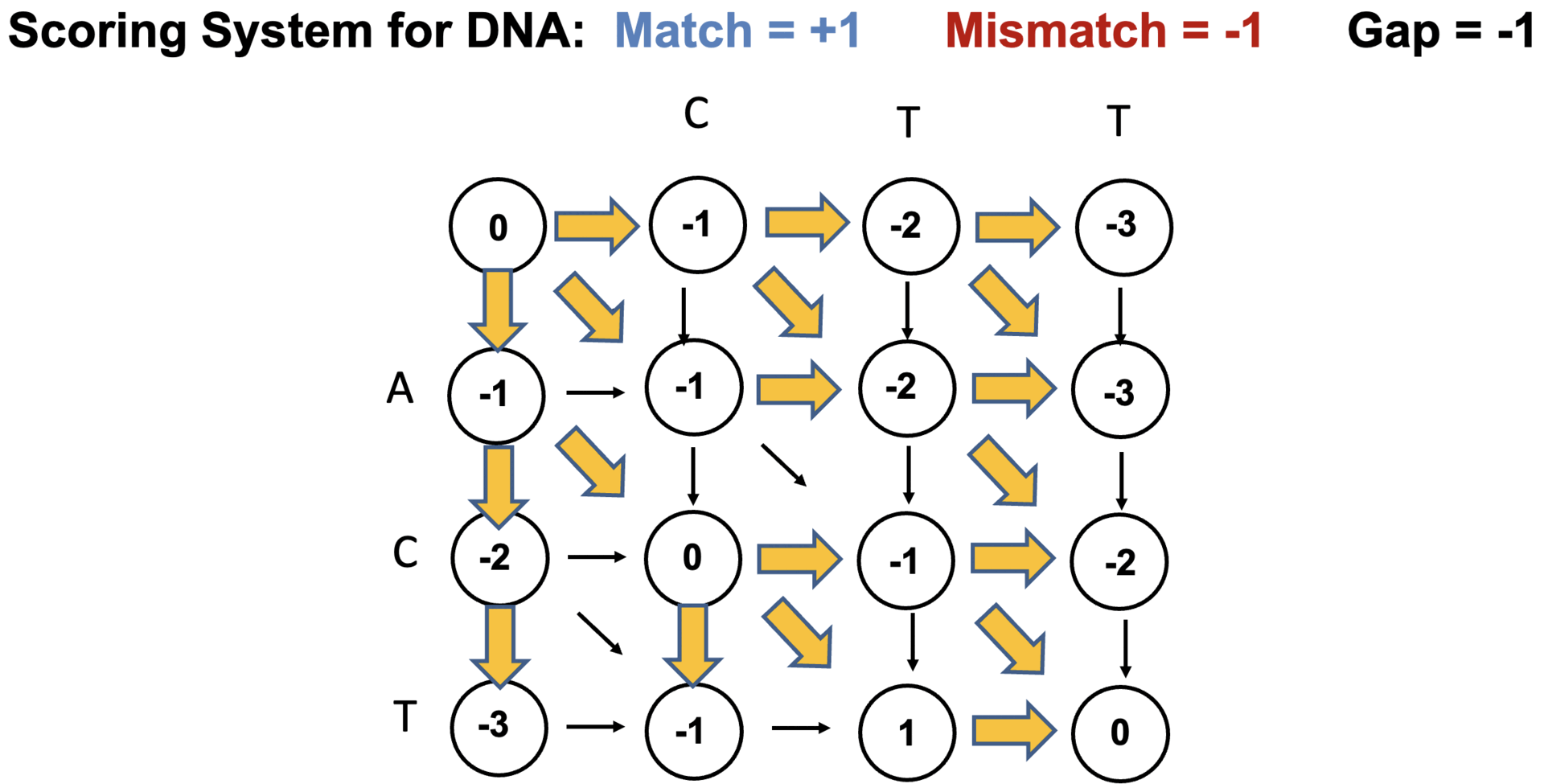
图 13. Needleman-Wunsch 图的构建
○ 第 2 阶段。 最优图路径搜索:动态规划。沿着最终节点(即 T-T)处的高值进行回溯。
图 14. Needleman-Wunsch 图中的最佳图路径搜索
○ 伪代码
○ 2-3. Smith-Waterman 图:配对对齐
○ 阶段 1. 构造局部对齐图:在全局对齐图中将零分配给负值。
○ 阶段2. 最优图路径搜索:动态规划。从具有最大值的节点回溯。
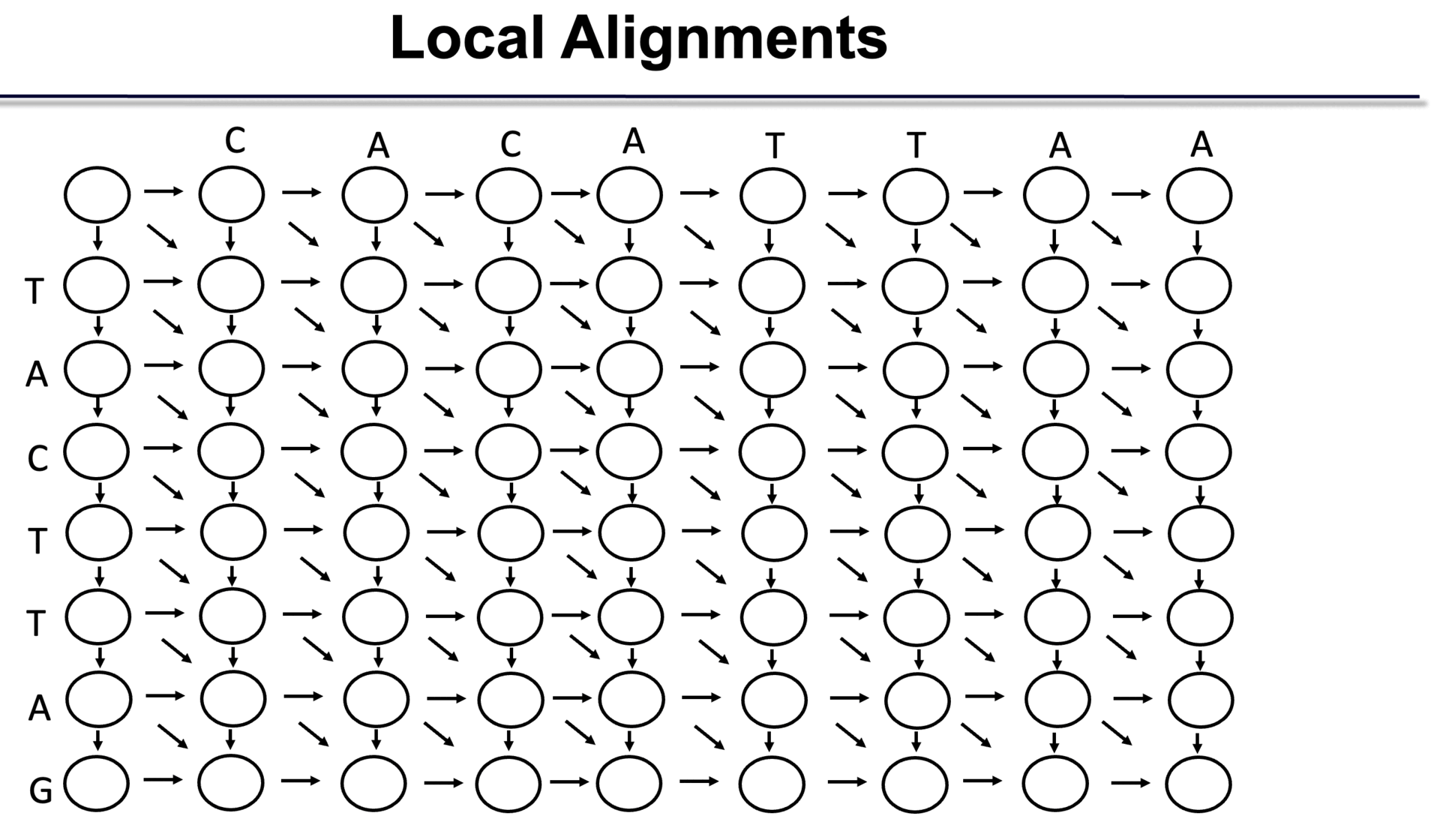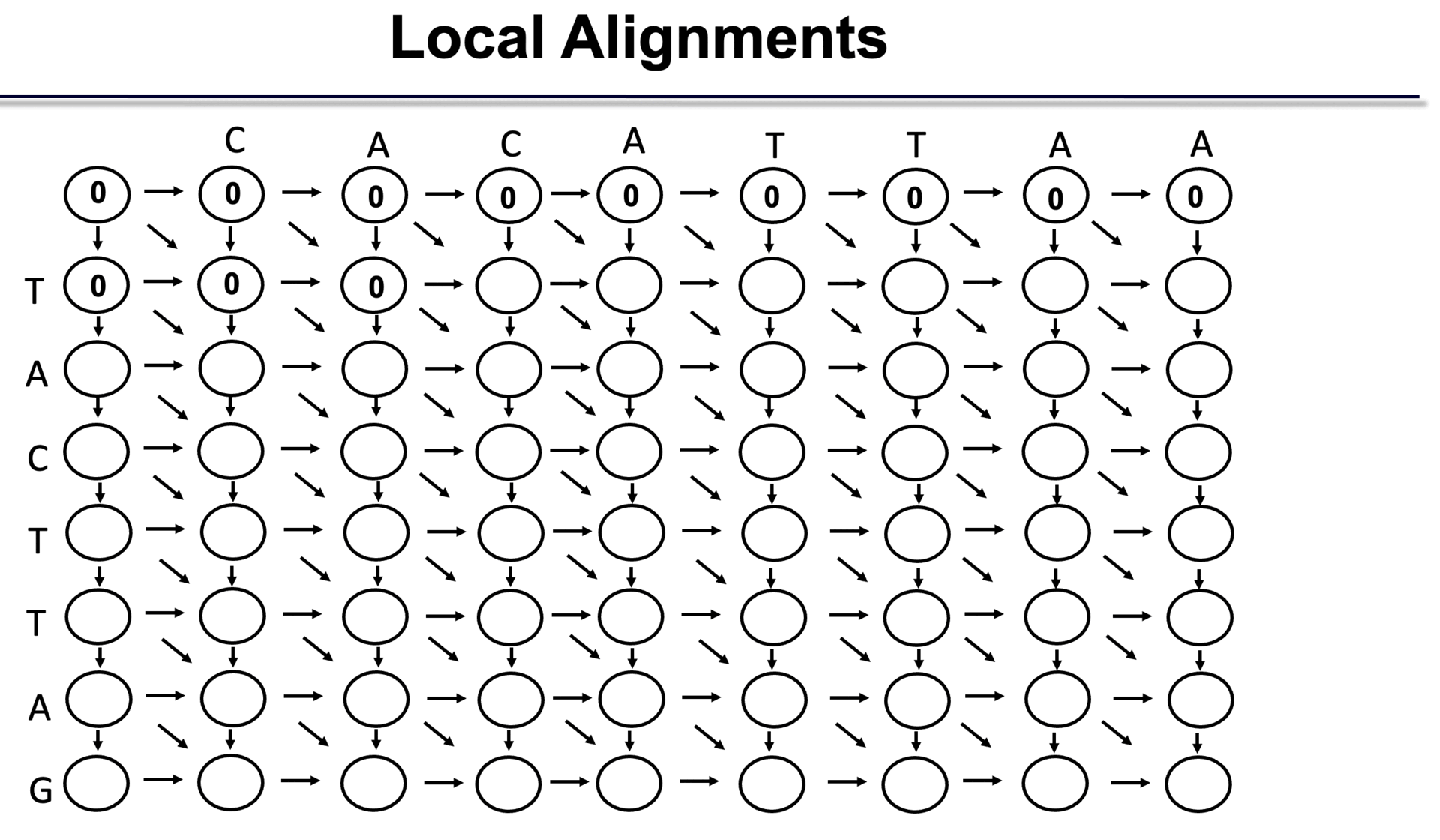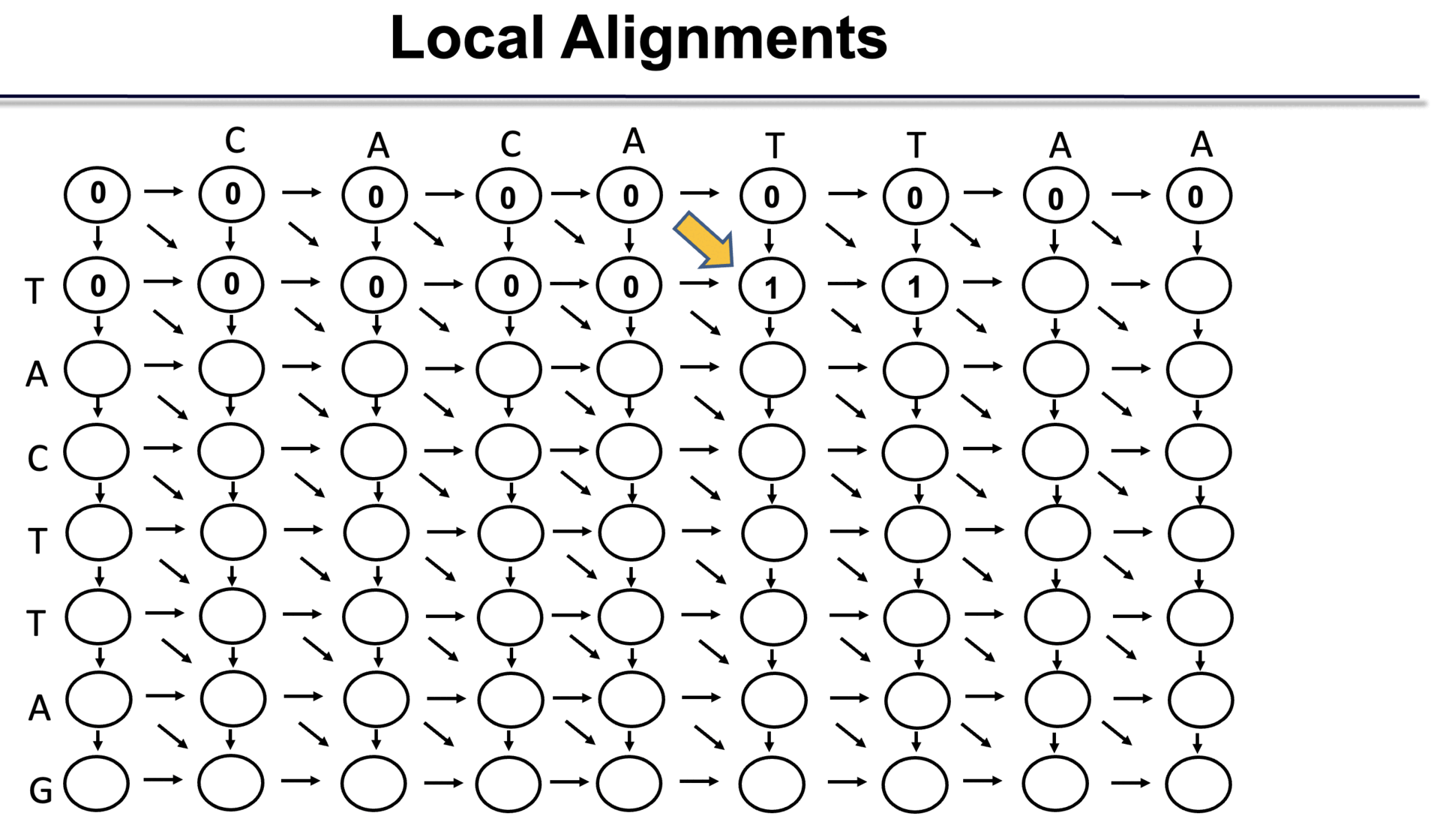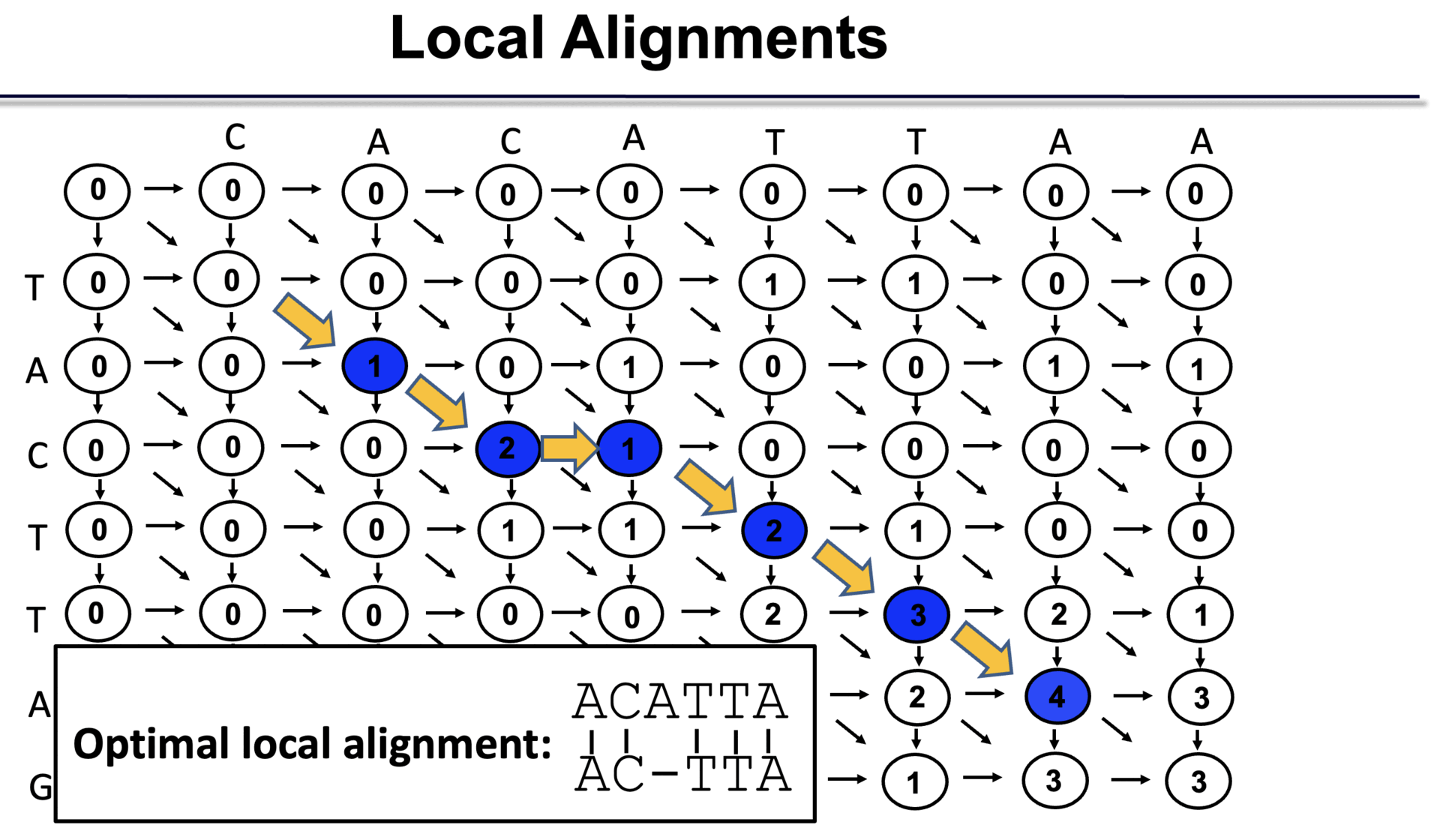
图 15. Smith-Waterman 图
○ 伪代码
○ 示例:如果出现差距,可能需要重新考虑。感觉序列分配顺序应该稍微延迟一下。
图 16. Smith-Waterman 图示例
○ 2-4. 渐进比对:MSA(多序列比对)
○ 使用基于树的策略,其中相似的序列最初进行比对,然后将其他序列与这些比对进行比对。
○ 在渐进式多序列比对中,序列比对的顺序很重要。
○ 2-5. 迭代比对:MSA(多序列比对)
○ 与渐进式类似,但进一步将初始序列重新调整为当前完整的 MSA。
○ 减少对初始对齐的依赖。
○ 2-6. 隐马尔可夫模型:MSA(多序列比对)
○ 可以利用 Profile HMM 来训练和迭代比对序列。
○ 2-7. UPGMA(算术平均的未加权配对法):MSA(多序列比对)
○ 第一第一。通过将所有可能的序列对对齐在一起来计算它们之间的成对编辑距离 (Smith-Waterman / Needleman-Wunsch)
○ 第二第二。使用凝聚层次聚类之一创建层次顺序,UPGMA。
○ 第三rd。通过向上遍历树,逐步将序列(和比对)对齐在一起。
○ 步骤3. 图遍历:搜索算法
○ 欧拉路径:图中每条边恰好访问一次的路径。如果存在重复序列,则两个节点可以通过多条边连接。不需要遍历整个图。广度优先搜索(BFS)和深度优先搜索(DFS)都是可能的。
○ Euler-SR
○ Velvet:对于中小型基因组快速,约 50 Mb 或更小。 Velvet 中的“Tour Bus”算法利用 Dijkstra 算法,使用覆盖率、序列标识和长度阈值消除图中的“气泡”。
○ 黑桃
○ 皮隆»» ○ Abyss:对于中小型基因组来说速度很快,约 50 Mb 或更小。
○ SSAKE (Warren et al. 2007):短读汇编器
○ VCAKE (Jeck et al. 2007):短读汇编器
○ miniasm:长读序列汇编器。使用来自多个映射的核苷酸序列中的距离来形成相应的边缘。
○ 在这种情况下,可以说查询序列的 ABCD 对应于目标序列 3 的 ABCD。
○ 步骤4. 清理装配图:如果对应的截面是P → Q → R,则可以简化为P → R。
○ 步骤 5. 生成单位。
○ 在此过程中,会产生位置、不匹配等信息。
○ 汇编过程的结果生成一个
.bam文件。
○ 第 3 类. 组合策略
○ 如果参考基因组质量较差或来自远缘物种。
○ 类型 1. wgs-汇编器
○ 类型 2. Falcon
○ 类型 3. ra-integrate
○ 类型 4. miniasm(一种用于长读测序的组装算法)
○ 类型5. 绒:用于了解断点连接序列的坐标和特征。
○ 类型 6. SPAdes:基因组组装
| 汇编器 | 从头开始? | 并行度 | 支持双端读取吗? | 支持搁浅读取吗? | 支持多种插入尺寸? | 输出转录本计数? |
|---|---|---|---|---|---|---|
| G-Mo.R-Se | 没有 | 无 | 没有 | 没有 | 没有 | 没有 |
| 袖扣 | 没有 | 国会议员 | 是的 | 是的 | 是的 | 是的 |
| 经文 | 没有 | 无 | 是的 | 是的 | 是的 | 是的 |
| 埃兰格 | 没有 | 无 | 是的 | 是的 | 是的 | 是的 |
| 多重 k | 是的 | 无 | 是的 | 是的 | 是的 | 没有 |
| 注释器 | 是的 | 国会议员 | 是的 | 是的 | 是的 | 是的 |
| 跨ABySS | 是的 | MPI | 是的 | 是的 | 没有 | 是的 |
| 绿洲 | 是的 | 国会议员 | 是的 | 是的 | 是的 | 没有 |
| 三位一体 | 是的 | 国会议员 | 是的 | 是的 | 没有 | 是的 |
表 3. RNA 组装的类型
⑤ 流程5. 共识完善
○ 类型 1. Quiver
○ 类型 2. 纳米抛光
⑥ 应用1.参考匹配» ○ 定义:当使用不同物种的多个参考文献时,确定读数来自哪个参考文献的过程。
○ 采用特殊算法来纠正由于种间同源性而产生的错误。
○ 通常,映射到不同参考文献的多个
.bam files用于为每个转录本查找更准确的参考文献。
○ 这一讨论意味着,为了准确、全面地绘制给定组织的 RNA 图谱,应该考虑一种或多种参考的最佳组合。
○ 类型 1. 单独对齐
○ 1-1. Freemuxlet:使用 SNP 对来自多个个体的细胞进行解复用。
○ 类型 2. 异种移植比对:使用移植物和宿主参考中的 .bam 文件匹配每个转录本的参考。
○ 2-1. XenofiltR:从主机和移植引用中获取 .bam 文件。生成
.bam和.bam.bai files,仅剩下移植读数。
○ 2-2. BAMCMP:分离异种移植数据中的移植和宿主读数。分为仅移植类、仅宿主类、模糊类、未映射类。
○ 2-3. 消除歧义
○ 2-4. Xenomake:仅用于空间转录组学。
○ 类型 3. 在伪对齐中使用多个参考(ref)
○ 构建集成参考(例如单个物种参考)的简单方法会导致准确性较低。
○ 类型 4. 微生物组排列
○ 细菌参考文献:SILVA、RDP(核糖体数据库项目)、Greengenes、RefSeq
○ 真菌参考文献:UNITE、EUKARYOME
○ 4-1. BLAST:慢但最准确
○ 4-2. VSEARCH:现在很少使用
○ 4-3. MAFFT:用于 MSA(多序列比对)
○ 4-4. DECIPHER:用于 MSA(多序列比对)
○ 4-5. Pathseq:使用 GATK(基因组分析工具包)对混合人类微生物宏基因组数据进行过滤、比对和丰度估计。
○ 4-6. Kraken
⑦ 应用2. 工作流程构建
○ 类型 1. SnakeMake (Python):用于独立创建自定义工作流程的工具。
○ 类型 2. SpaceMake:处理各种空间转录组数据的集成工作流程。
○ 类型 3. Nextflow (Java)
○ 类型 4. Galaxy (Python)
○ 类型 5. waf (Python)
○ 类型 6. SCons (Python)
○ 类型 7. 耙子(红宝石)
○ 类型 8. BioMake(现为 Skam)(Prolog)
○ 类型 9. Ruffus (Python)
○ 类型 10. 摊铺机 (Python)
○ 11 型. 安杜里尔
○ 类型 12. bcbio-nextgen (Python)
○ 类型 13. gkno (Python)
○ 类型 14. 调用 (Python)
○ 类型 15. 队列(Scala、Java)
○ 类型 16. NGSANE(bash)
○ 类型 17. BigDataScript (bds)
○ 类型 18. Bpipe (Groovy)
○ 类型 19. Omics 管道 (Python)
○ 类型 20. Cromwell/WDL (Scala)
○ 类型 21. Toil (Python)
⑷ 步骤 3.(可选)QC
① SAMtools: 可以使用 SAMtools 视图。可以通过标志进行过滤。 ([参考](https://broadinstitute.github.io/picard/explain-flags.html))
⑸ 步骤4. 排序:沿对齐轴对BAM文件进行排序。> ① Picard (release, 麻烦- 拍摄)
受保护_5
② SAMtools
受保护_6
③ name-sorted:通常,根据读取的名称进行排序。
④ 坐标排序:例如,在SpaceRanger 中创建的
possorted_genome_bam.bam file是位置排序的。
⑹ 步骤 5. 标记: 标记 PCR 扩增产生的重复项。
① 一般来说,DNA-seq涉及去除重复,而RNA-seq则不会:在RNA-seq中,相同的序列可能会重复出现,不仅是由于技术上的重复,还因为高表达的转录本或短基因。删除这些生物重复可以减少数据的动态范围或降低统计功效。
② Picard MarkDuplicates
受保护_7
③ SAMtools rmdup
受保护_8
○
-s:单端读取。如果是双端读取,则去掉这部分。
④ 修剪机
⑤ 修剪丰富!
⑥ 快速
⑺ 步骤 6. 索引: .bam.bai file 是从 .bam file 生成的。
① SAMtools
受保护_9
⑻ 步骤7. 计数: 当有read时,确定read源自哪个特征(例如基因、外显子)。
图 17. 计数过程(来自 HTSeq)
① 当有read时,判断read来自A基因还是B基因的算法。
② 类型 1. HTSeq
○ 基于基因重叠的工具。
○ 最常用计数算法。
○ 默认为并集。
○ 命令示例:
受保护_10
○ -f bam:指定输入文件是.bam file。
○
-r pos:确保输出文件中的特征顺序与参考中的顺序匹配。
○
-s no:-s表示测序数据是否是链特异性的。-s no指定数据不是特定于链的,这意味着不考虑读取的转录方向(链)。对于特定于链的数据,您应该使用-s yes(正向链)或-s reverse(反向链)。
○
-i gene_id:使用 GTF 文件中的gene_id 属性作为每个特征的参考。 transcript_id 也是可能的。
○
-t exon:指定要素类型。默认为外显子。
○
Mus_musculus.GRCm38.99.gtf:参考名称。可以使用.gff文件代替.gtf。
○
> output.txt:将计数结果输出到output.txt 文件。
○ 结果示例:
○ 简单的计数算法,无需组装过程。
○ 问题 1. 当存在多个同工型时,共享读段要么一致计为 +1,要么被丢弃,导致计数不准确。
○ 问题 2: 与基因水平相比,转录水平计数的准确性降低
○ 基因计数 > 构成每个基因的异构体计数总和(∵ 模糊读段)
○ 使用基因组比对数据进行转录本比对而不进行单独组装可能不准确
○ 由于转录本之间高度相似,通过测序技术进行区分可能极具挑战性
③ 类型 2. RSEM» ○ 利用 EM(期望最大化)技术准确预测计数。
○ 可以进行异构体定量。
○ GDCPortal 提供 RSEM 计数值。
○ 步骤 1: RSEM 安装
受保护_11
○ 第2步: 准备参考文件
受保护_12
○ 步骤 3: 表达估计
受保护_13
④ 类型 3. StringTie
受保护_14
○ 基于基因组:(评论)StringTie 的转录水平丰度方法并不简单
○ 包括组装过程。
○ 将基因组比对读取中的
sorted_bam.bam file作为输入,输出单独的output.gtf file。
○ 采用新颖的网络流算法
○ 结果示例
⑤ 输入 4. featureCounts (安装)
受保护_15
○ 结果示例
⑥ 类型 5. 袖扣 / Cuffdiff
⑦ 类型 6. Tuxedo:可以进行异构体定量。
○ 步骤 1. 袖扣:组装每个样本的转录本,并用最少数量的亚型解释观察到的读数。
○ 步骤 2. 合并 GFT 文件。
○ 步骤 3. Cuffquant:进行转录本定量。
⑧ 类型 7. Hisat2:基于基因组
⑨ 类型 8. QoRT
⑩ 类型 9. eXpress
○ 利用 EM(期望最大化)技术。基于转录组。
○ 可以进行异构体定量。
⑪ 类型 10. bowtie2:基于转录组
⑫ 11 型. TIGAR2
○ 用于转录本量化的贝叶斯推理。
○ 可以进行异构体定量。
⑬ 类型 12. 鲑鱼
○ 准作图,而非碱基对碱基比对 + 异构体定量
○ 对于基因水平分析,使用 tximport。
○ 步骤 1. 从头开始或从头组装
○ 步骤 2. 构建哈希表:对不同 k 聚体的位置进行索引。
○ 步骤3. 构建后缀数组:注释k-mers的后缀。
○ 步骤4. 准映射
○ 4-1. 从左到右扫描read:扫描直到在哈希表中找到k-mer。识别检测到的 k 聚体的后缀。
○ 4-2. 识别MMP(最小映射位置):找到精确匹配的最长匹配读序列,以确定MMP。
○ 4-3. 假设存在不匹配,识别 NIP(下一个位置):通过跳过一个 k 聚体来识别 NIP,从而解释读数中的测序错误或自然变异。不要在当前匹配中断的地方停止,而是搜索下一个匹配的 k-mer,以增加映射整个读取的可能性。
○ 4-4. 重复步骤4-1和4-2,直到到达read末尾,完成准映射。
○ 步骤 5. 使用 EM(期望最大化)算法量化转录本丰度。
○ 第 1 步: 准备
.ctab文件
○ 方法一: TopHat2 + StringTie
○ 方法2:礼帽2 + 袖扣 + Tablemaker
○ StringTie 命令示例:使用 -B 参数生成 .ctab 文件
受保护_16
○ 第2步:检查目录结构
受保护_17
○ 第3步: 执行 Ballgown:可以使用 R 运行
受保护_18
○ 结果示例> ⑮ 类型 14. Pathseq:使用 GATK(基因组分析工具包)对人类和微生物的混合宏基因组数据进行过滤、比对、丰度估计。
⑯ 类型 15. IUTA(异构体使用两步分析):异构体定量
⑰ 类型 16. CellRanger
○ scRNA-seq 比对和计数
⑱ 类型 17. SpaceRanger
○ ST 对齐和计数
○ 仅限 Visium。使用 STAR 对齐并另外考虑空间条形码
⑲ 类型18. TE(转座因子)定量
○ scATAC-seq:床具
○ scRNA-seq 和幻灯片-seq:kallisto
○ snmC-seq:所有COOls
⑼ 步骤 8. (可选) 纠错:可以在组装过程中纠正映射错误。
① pbdagcon
③【纳米正确】(http://www.ncbi.nlm.nih.gov/pubmed/26076426)
④ 打火机:基于k-mer。基于C++。
⑽ 步骤 9.(可选)变异识别:可以研究遗传变异,如 SNP 或插入缺失。变体保存为 VCF 文件。
① 概述:典型的人类基因组与参考人类基因组有 410 万至 500 万个位点不同
○ 大多数 (>99.9%) 是 SNP 和短插入缺失。
○ 包含 2,100 至 2,500 个结构变体,影响约 20 Mb 的序列,并包含(大约):
○ 1000 个大插入缺失
○ 160 拷贝数变异
○ 915 个 Alu 插入件、128 个 L1 插入件、51 个 SVA 插入件
○ 4 个坚果
○ 10 次反转
○ 单个基因组中的大多数变异都是常见的。
○ 每个基因组中只有 40,000 到 200,000 个 (1-4%) 的频率 <0.5%。
② 管道
○ 方法:读段对映射、读段深度分析、拆分读段比对、序列组装
图 18. 变体调用方法
○ 随后创建了BEDgraph文件(由BED组成的图形数据结构文件)、Wiggle文件(与对照组比较的文件)和bigWig文件 (Wiggle 文件的压缩二进制版本)。
③ 类型
○ GATK(Genome Analysis ToolKit):常用的是GATK中的HaplotypeCaller。此外,还有 UnifiedGenotyper 和 Mutect2。
○ Freebayes
○ SAMtools 编译
○ CaVEMan(通过期望最大化的癌症变异):用于体细胞替代调用。
○ Pindel:用于 Indel 调用。
○ BRASS (BReakpoint AnalySiS):用于结构变异调用。
○ SPP
○ GWAVA(全基因组变异注释)
○ DeepSea:预测非编码变体
○ DanQ:使用 CNN 和 RNN 量化 DNA 序列的功能
○ DeepFun:使用 CNN 预测监管变体
○ DeepC:预测 3D 基因组折叠
○ 秋田:预测 3D 基因组折叠» ○ 津巴猫
○ 胡椒
○ ANNOVAR:变异注释(编码/非编码/染色质标记)
○ VEP:变异注释
○ SnpEff:变异注释
○ 普联
○ LD(连锁不平衡)计算
○ 遗传相关性计算:如果存在遗传相关性,遗传独立性就会受到损害,导致有效大小的高估或显着性的夸大。
○ 支持多种输入输出格式(包括VCF/剂量估算数据)
○ 常用格式:
○
.bed(存储基因型矩阵的二进制文件)、.fam、.bim:全部具有相同的前缀
○
.pgen(二进制文件)、.psam、.pvar
○ vcftools
○ 设计用于处理测序项目生成的 VCF(变体调用格式)文件。
○ 可以输出为PLINK格式。
④ 应用1. 基因型聚类
○ GenCall(来自 Illumina 的 GenomeStudio)
○ 样本间模型,一次一个标记
○ 将基因型分配给最近的簇
○ 可以自定义先前的集群
○ 基因SNP
○ 样本内模型,样本中的所有标记
○ 使用变分贝叶斯 EM 模型
○ 可以很好地处理稀有变体
○ 可以并行
○ 光学呼叫
○ 样本间和样本内模型的混合
○ EM 算法来拟合 t 分布的混合
○ zCall
○ 后处理工具
○ 根据常见变异的纯合子簇的均值/方差划分 X/Y 坐标,然后回忆罕见变异
⑾ 步骤 10.(可选)事后 ** **转录物到基因的转换:当有转录物 ID 的计数数据时,可能需要将多个转录物 ID 折叠成一个基因 ID(例如,用于 GO 分析)
① 示例:GRCh38(人类)GFF 文件包含以下信息
受保护_19
② 转换后方法
○ 方法 1. RefSeq
○ 方法 2. UCSC已知基因
○ 方法 4. GENCODE
○ 方法 5. R 中可用的有用函数(对于人类,ref)
受保护_20
5。 QC 5. 标准化
⑴ 概述
①定义:纠正由于技术限制RNA读数不能准确反映基因表达的偏差
② 本质上是纠正系统批次效应,例如文库大小
⑵ 类型1.文库大小标准化(基于深度的标准化)
① 比较不同样本时,将每个样本除以归一化因子以调整总 RNA 转录本计数
○ 换句话说,比较样本之间的特定基因表达
○ 测序深度是测序机特有的限制,可以称为文库大小
② 1-1. RPM(每百万映射读取的读取数) 或 CPM(每百万映射读取的计数)
○ 将每个基因计数除以样本总数,然后乘以 10^6
○ R代码:保持相同的库大小,不使用TMM等方法
受保护_21
> ③ 1-2. TMM(M 值的修剪平均值)(参考)
○ 定义:通过除以样本的总读取计数+修剪来标准化给定的读取计数
○ Robinson 和 Oshlack 提出 (2010) (参考)
○ 由于执行了修剪,库大小不会保持恒定。
○ 显着性 1. 纠正读数计数不能准确代表实际基因活性且与测序深度成正比的偏差(参考)
○ 条件: 确保两个样本中表达水平相同的基因不会被检测为 DEG
○ 假设: 大多数基因没有差异表达(与作者的经验一致)
○ 思想实验
○ 样品 A 是人 + 小鼠 RNA 的混合物,样品 B 是样品 A 中的人 RNA,两个样品具有相同的人和小鼠 RNA 计数
○ 当深度相等时,A 中的人类 RNA 读数将恰好是 B 中的人类 RNA 读数的一半:A 中的读数分布在基因上的两倍
○ 要调整 A 中每个基因的 RNA 读取计数,请乘以 B 的两倍的因子(归一化因子)
○ 这个假设导致批次+样本效应≃批次效应,使得归一化方法仅适用于这种情况
○ 意义 2. 确定总 RNA 产量 Sk 很困难,但计算两个样本的比率 Sk / Sk’ 相对容易
○ 简要定义:将给定的读取计数除以样本的总计数
○ 示例
○ 下图为样本1与样本2的g基因表达差异
○ 第一第一。符号定义
○ Lg: 基因g的长度
○ μgk:样本k中基因g的实际转录本计数。代表表达水平。与人口相关
○ Nk:样本 k 中的转录本总数
○ Sk:样本 k 中的 RNA 计数
○ Sk / Nk:每个转录本的平均 RNA 计数
○ Ygk:观察到的样本 k 中基因 g 的转录本计数。与样本人群相关
○ Mg:基因对数倍数变化
○ Ag: 绝对表达水平
○ Sk / Sr:样本 k 中除以的缩放因子。与 Sk 成正比。参考上面的思想实验。
○ TMM:归一化因子
○ 第二第二。删除表达量为0的基因
○ 第三第。修剪:与 RPM 或 CPM 最根本不同的方面。
○ 截尾平均值:排除顶部 x% 和底部 x% 的数据平均值»> ○ 双重修剪:根据对数倍数变化 Mrgk 和绝对强度 Ag 进行修剪
○ 最初的研究人员通过 Mrgk 削减了 30%,通过 Ag 削减了 5%
○ 第 4。除以样本 k 的 TMM
○ wrgk:低表达基因的权重较大,以防止失真
○ 当 Nk = Nk’ 且 Ygk = 2 × Ygr 时,TMM 约为 2
○ R代码:用TMM更改库大小,然后计算CPM
受保护_22
○ 应用1. GeTMM方法
④ 1-3. RLE(相对对数估计)
○ 由 Anders 和 Huber (2010) 提出。
○ R包DESeq和DESeq2中默认采用的归一化方法。
○ 步骤 1. 创建伪参考(中值库):对所有样本取几何平均值。
○ X:原始计数
○ g:基因
○ k: 条件
○ r:复制
○ 步骤2. 每个样本与伪参考的中值比率用作比例因子(尺寸因子)。
○ 步骤 3. 该样本的基因计数值除以比例因子被视为该基因的标准化计数。
○ R代码
受保护_23
⑤ 1-4. UQ(上四分位数)归一化
○ 一种在分析两个转录组时将特定分位数的表达水平调整为相同的方法。
○ Bullard 等人提出。 (2010)。
○ 通常使用上 75%(下 25%)四分位值作为变量(参见 Q3-范数)。
○ 主要用于微阵列数据。
○ R 代码
受保护_24
○ 考虑空间区域特定文库大小偏差的文库大小标准化
○ 基于负二项分布
⑶ 类型2. 基因长度标准化
① 定义:比较单个样本内不同基因(或外显子、亚型)表达时校正基因长度的方法。
○ 这里的基因长度是指有效长度,即实际基因长度减去读长。
○ 基因长度校正:基因计数与基因长度成正比,因此计数值除以基因长度。
○ 基因长度归一化因不能正确反映数据特征而受到批评,FPKM、TPM 等指标的重要性不断下降。
② 2-1. RPKM(每百万映射读取中每千碱基转录本的读取数)
○ 公式:基因 i 的 RPKM 表示为 Q 个读数和ℓ 外显子长度。
○ 用于比较样本中不同基因之间的基因表达。» ○ 示例: 基因中的 25,000 个读数 / (0.5 kb 基因 × 4000 万个读数) = 1,250
③ 2-2. FPKM(每百万映射片段中每千碱基外显子的片段)
○ 公式:基因 i 的 FPKM 表示为 q 个读数和 ℓ 外显子长度。
○ 片段是指双端测序中的一对reads。
○ 与 RPKM 类似,但仅用于双端 RNA-seq (Trapnell et al., 2010)。
○ 示例: 基因中 25,000 个配对末端片段 / (0.5 kb 基因 × 4000 万个配对末端读数) = 1,250
④ 2-3. TPM (Transcripts Per Kilobase Million):由 Li 等人提出。 (2010)
○ 定义:根据样本中每个 1M RNA 分子有多少个来自该基因的分子进行标准化。
○ 与 RPKM 的唯一区别:在计算 TPM 时,首先对基因长度进行归一化,然后对测序深度进行归一化。
○ 由于基于长度的标准化,相对易于使用,比 CPM 更常见。
○ 将单个库中的 TPM 值相加得到 1,000,000,允许不同样本之间的比较。
○ TPM 和 RPKM 之间的关系: 对于基因数 n,
○ TPM 与 RPKM(或 FPKM)结果高度相关
○ 仍然存在一些偏差,例如测序深度和潜在的技术工件。
⑤ 2-4. ERPKM
○ RPKM 的改进,用有效读长(即基因长度 - 读长 + 1)代替基因长度。
⑷ 类型3. 未知技术工件标准化
① RUV(去除不需要的变异)
② SVA(替代变量分析)
③ PCA(主成分分析)
图 19. 各种归一化方法
⑸ 类型4. 对数转换
① 目的1: 对于计数值较高的基因,这些基因的实际活性在解释时可能会被夸大。
② 目的2: 当同时存在非常小和非常大的值时,需要调整尺度使其相似。
③ 针对上述问题,将归一化计数进行对数转换的结果视为基因表达值。
④ 变换有多种,例如幂变换,但对数变换是最先进的(SOTA)(ref)。
⑹ 类型 5. 缩放(例如,z 分数变换)
① 对于每个患者和每个样本,基因表达值的范围各不相同。为了合理比较,对取值范围进行了调整。
表 4. 各种缩放方法
⑺ 类型6. 特征选择、单元格选择
⑻ 类型 7. Lambda GC:SNP 数据标准化方法
① 步骤1. 分析每个SNP 与表型之间的相关性。
② 步骤 2. 将 p 值转换为 z 分数。例如,在 R 中:
z = qnorm(p / 2)
③ 步骤3. 计算卡方统计量。例如,在 R 中:
c = z^2> ④ 步骤 4. 计算所有 SNP 的卡方统计数据的中位数。
⑤ 步骤 5. Lambda GC:将上述中位数除以 0.455(即 df = 1 时 χ2 的中位数)。期望值为 1。
⑥ 步骤 6. 将每个 SNP 的实际 χ2 值除以 lambda GC,然后反向计算 p 值。
⑼ R:使用Seurat包获取标准化表达。
受保护_25
⑽ Python:使用scanpy包获取标准化表达式(ref)
受保护_26
⑾ 论文表述
○ 通过每百万计数 (CPM) + 1 个伪计数的 log2 转换对计数数据进行归一化,并使用 R Studio(版本 1.1.453)中的 ggplot2 包 (28) 为每对连续部分生成散点图。内置统计包用于计算 Pearson 相关性。 ([参考](https://aacrjournals.org/cancerres/article/78/20/5970/631815/Spatially-Resolved-Transcriptomics-Enables))
6。 QC 6. 批量效果
⑴ 概述
① 背景:除了实验变量外,批次效应也会影响实验组和对照组之间的结果。
② 批次效应:实验结果受生物变量以外的因素影响。示例包括:
○ 排序日期
○ 研究人员进行测序
○ 测序设备
○ 协议
③ 诸如文库大小之类的系统批次效应可以通过归一化来纠正。
④ 批次效应去除过程简称为去除非生物批次效应,常采用回归分析。
⑵ 批量效应去除
① 这对于通过样本之间的比较进行 DEG 分析尤为重要。
② 注意事项
受保护_27
○ 如果特定条件适用于特定批次,则理论上无法消除批次效应。
○ 但是,即使在这种情况下,也可以通过区分批次效应和协变量来创建不完全回归模型以消除批次效应。
③ 方法一: limma::removeBatchEffect:通过将归一化表达矩阵和批次信息输入线性模型来去除批次效应。 ([参考](https://rdrr.io/bioc/limma/man/removeBatchEffect.html))
④ 方法 2: sva::ComBat (Johnson et al., 2007):基于经验贝叶斯的方法,用于在批次信息已知时消除批次效应。 ([参考](https://www.bioconductor.org/packages/release/bioc/vignettes/sva/inst/doc/sva.pdf),[参考](https://scanpy.readthedocs.io/en/stable/api/scanpy.pp.combat.html),[参考](https://academic.oup.com/biostatistics/article/8/1/118/252073))。使用线性模型和经验贝叶斯收缩。
受保护_28
⑤ 方法 3: ComBat_seq (Zhang et al., 2020):采用原始计数矩阵并输出调整后的计数矩阵。应了解批次信息。
受保护_29
⑥ 方法 4: SVA seq (Leek, 2014):即使没有已知批次信息也能发挥作用。
⑦ 方法 5: RUV seq (Risso et al., 2014)
⑶ 批量效应去除精度的测量
① k-近邻批量效应检验(kBET)
② 跨批次反潜战
③ k近邻(kNN)图连通性
④使用PCA回归批量去除
⑷ 应用一:聚类与批量效应
① 聚类可用于消除批次效应,通过在主要簇(例如细胞类型簇)内划分实验组和对照组来获得 DEG。
⑸ 应用二:数据集成
① 合并不同批次或方式的数据,并考虑批次修正效果。
② 修改两个组,使其具有相同的特征。» ○ 示例 1: 训练模型,使聚类模式不受批次效应的影响。
○ 示例 2: 训练模型以最小化鉴别器的性能,该性能根据批次特定的效果来区分数据(即,使数据在集成后无法区分)。
图 20. 数据集成类型
④ 方法 2. Seurat V1 和 V2:使用 CCA(典型相关分析)
○ 具有用于存储原始计数或标准化数据的表达式值的单个数据槽。
○ 主要为单一模式设计。
○ 如果批次效应较大或共同细胞亚群较少,则整合效果不佳。
○ 基于 CCA 的数据集成:此方法积极地将两个组对齐。
○ 基于 RPCA 的数据集成:此方法对两组进行不太积极的对齐,这在两个数据集的组织特征显着不同时非常有用。
⑤ 方法3. Seurat V3
○ 步骤 1. 使用对角化 CCA 对参考数据集和查询数据集执行降维。
○ 步骤 2. 将 L2 范数应用于规范相关向量。
○ 步骤 3. 计算 MNN(相互最近邻)以探索两个数据集共享的低维表示:这涉及识别和链接数据集中具有相同上下文的单元格。这些链接的单元对称为锚点。
○ 步骤 4. 对锚点进行评分并删除错误链接的低置信度锚点:分数定义为共享邻居重叠。
○ 步骤 5. 定义加权距离:给定第 i 个锚点 ai、查询单元 c 和锚点得分 Si,
○ 步骤 6. 对加权距离应用高斯核:默认标准差 (sd) 为 1。
○ 步骤 7. 标准化所有 k.weighted 锚点。
○ 步骤 8. 计算所有锚单元对的积分矩阵 B = Y[:, a] - X[:, a],其中 X 是参考表达矩阵,Y 是查询表达矩阵。
○ 步骤 9. 使用权重矩阵 W 和积分矩阵 B 计算变换矩阵 C。
○ 步骤10. 将原始查询表达矩阵Y减去变换矩阵C,得到整合表达矩阵。
○ 步骤 11. 标签传输:使用二值化的锚分类矩阵 L 和权重矩阵 W 计算标签预测 Pl。
⑥ 方法 4. Seurat V5
○ 具有快速、更高效的锚定以及改进的内存处理能力。
○ 不仅可以集成测量核酸的模式,还可以集成测量蛋白质等其他模式。
⑦ 方法5. BBKNN(批量平衡k-近邻):使用PCA
⑧ 方法6. mnn正确
⑨ 方法7. Scanorama:在Python scanorama中使用。使用奇异值分解
⑩ 方法 8. Conos :在 R conos 中使用。
⑪ 方法 9. scArches:利用迁移学习。
⑫ 方法10. DESC
⑬ 方法 11. fastMNN (batchelor):使用 PCA
⑭ 方法 12. Harmony:使用 PCA。 Harmony 不提供批量效应校正的归一化表达,但它确实提供了可用于下游分析的批量效应校正的低维嵌入。
⑮ 方法 13. LIGER:整合单细胞转录组、表观基因组和空间转录组数据的通用方法。基于因素。使用积分非负矩阵分解
⑯ 方法 14. SAUCIE
○ 优点:提供批次效应校正的标准化表达,并包含用于识别差异表达基因 (DEG) 的参考代码。 ([参考](https://www.kaggle.com/code/hiramcho/scrna-seq- Differential-expression-with-scvi#4.-Differential-EXpression))
○ 缺点 1. 不适用于表观遗传学数据(例如 scATAC-seq)。
○ 缺点2. 单独提供的DEG识别码速度慢且不准确。由于所有 10 个数据集和其中 2 个数据集之间的批次效应校正存在细微差异,DEG 结果有时可能会完全相反。
○ 建议:建议使用scVI标准化表达和
scanpy.tl.rank_genes_groups进行DEG识别。
㉑ 方法19. TrVaep
㉒ 方法20. scIB:除了合并多个集成工具外,还提供了基准测试。
○ 集成功能:BBKNN、ComBat、DESC、Harmony、MNN、SAUCIE、Scanorama、scANVI、scGen、scVI、trVAE
○ 基准指标:ARI、ASW、F1、相互评分等。
㉓ 方法 21. iMAP
㉔ 方法 22. INCT
㉕ 方法 23. scDML
㉖ 方法 24. scDREAMER,scDREAMER-Sup:它甚至可以实现物种间整合。
㉗ 方法25. SATURN:不同物种数据集的整合> ㉘ 方法 26. GLUE (Cao and Gau, 2023), scGLUE, SpatialGlue:用于集成来自不同平台的不配对数据的工具,例如scRNA-seq、scATAC-seq 和 snmC-seq。
○ 步骤 1. 将来自每个组学的细胞 × 特征数据嵌入到细胞 × 嵌入中,其中每个细胞都以公共维度表示。
○ 步骤 2. 为了表示组学之间的关系,使用基于知识的指导图来表示调节组 × 嵌入,其中每个调节组表示为与 步骤 1 相同维度的向量。
○ 步骤 3. 通过将 步骤 1 和 步骤 2 的输入馈入编码器,构造一个自动编码器,解码器输出集成数据。
○ 步骤 4. 对抗性学习:引入判别器来识别特定于平台的效果,并搜索可最小化判别器性能的自动编码器参数(从而促进更好的集成)。
㉙ 方法 27. Seurat Anchor (Stuart et al., 2019):基于因素
㉚ 方法 28. DC3(Zeng 等人,2019):基于因子
㉛ 方法 29. 耦合 NMF(Duren 等人,2018):基于因子
㉛ 方法 30. SCOT (Demetci et al., 2022):基于拓扑
㉛ 方法 31. UnionCom (Cao et al., 2020):基于拓扑
㉛ 方法 32. Panoma (Cao et al., 2022):基于拓扑
㉛ 方法 33. MrVI
㉛ 方法 34. scGCN:支持多组学
㉛ 方法 35. scETM
㉛ 方法 36. MultiVI:空间多组学数据集成
㉛ 方法 37. 生物领主
㉛ 方法 38. AutoXAl40mics
㉛ 方法 39. OmicsPlayground
㉛ 方法 40. MaxFuse:scRNA-seq + 空间蛋白质组学
㉛ 方法 41.totalVI:空间多组学数据集成
㉛ 方法 42. scMM:空间多组学数据集成
㉛ 方法 43. MEFISTO:scRNA-seq 或 ST 整合。输入数据应全部为 scRNA-seq 或 ST。
㉛ 方法44. StabMap:空间多组学数据集成
㉛ 方法 45. MOFA+:空间多组学数据集成
㉛ 方法 46. Seurat WNN:空间多组学数据集成
㉛ 方法 47. scINSIGHT
㉛ 方法 48. 杰米
㉛ 方法 49. 帕莫纳
㉛ 方法 50. SIMBA
㉛ 方法 51. scMRDR
⑹ 2-1. scRNA-seq 或 ST 之间的合并
① Seurat:除聚类外,大多数方法都需要将DefaultAssay从Integrated切换为RNA/Spatial/SCT等
○ 查找标记
○ 查找所有标记
○ 特征图
○ 空间特征图
○ 点图
○ 暗淡图
○ 空间DimPlot
○ 基因相关
○ 轨迹分析
② BANKSY:实现空间多组学之间的数据集成
⑺ 2-2. 将 scRNA-seq 数据与其他模式合并
图 21. 合并 scRNA-seq 数据
表5. 匹配数据的分析方法
表6. 未匹配数据的分析方法
7. Common 1. 集群
⑴类型1:【K均值聚类】(https://jb243.github.io/pages/2150#footnote_link_67_51)
⑵ 类型2:【无监督层次聚类】(https://jb243.github.io/pages/2150#footnote_link_67_50)
⑶ 类型3:矩阵分解> ① 将已知矩阵 A 分解为 W 和 H 矩阵的算法:A ~ W × H
○ 矩阵:代表样本和特征。可以从样本中推断。
○ H矩阵:代表变量和特征。
○ 类似K表示聚类,PCA算法。
○ 自动编码器 是比矩阵分解更广泛的概念,因为它包含非线性变换。
○ 以下算法基于最小二乘法,但也可以利用梯度下降等方法。
②算法:R = UV,其中R ∈ ℝ5×4,U ∈ ℝ5×2,V ∈ ℝ2×4,搜索U和V。
受保护_30
③ NMF(非负矩阵分解)
受保护_31
④ 矩阵完成(Netflix 算法):对 masked R 执行矩阵分解。
受保护_32
⑤ 应用1. 细胞类型分类
○ 旨在从组织的 scRNA-seq 数据中获取细胞类型比例。
○ 对于减少细胞类型异质性造成的混杂影响非常重要。
○ 3-1. 约束线性回归
○ 3-2. 基于参考的方法
○ 3-2-1. CIBERSORT(通过估计 RNA 转录本的相对子集进行细胞类型识别):允许您检查每个样本的细胞类型比例和 p 值。 Nu-支持向量回归(NuSVR)是CIBERSORT的核心算法。
○ 3-2-2. 史诗
⑥ 应用2.联合NMF:扩展到多组学。
⑦ 应用3:元基因提取
⑧ 应用4. Starfysh
○ 以下算法从空间转录组数据推断原型并确定代表每个原型的锚点。
○ 它可以实现细胞类型或细胞状态 DEG 的无参考推断,并允许使用 H\&E 信息估计细胞类型分布。
○ 步骤1. 构造一个自动编码器
○ X ∈ ℝS×G:输入数据(点×基因)
○ D:原型数量
○ B ∈ ℝD×S:编码器。在推断原型的背景下,每个原型在所有点上的分布总和必须为 1。
○ H = BX:潜变量
○ W ∈ ℝS×D:解码器。在重建输入数据的情况下,每个点的所有原型的权重之和必须为 1。
○ Y = WBX:重构输入
○ 步骤2. 求解优化算法来计算W和B
○ 步骤 3. 选择 W 矩阵中每个原型具有最高权重的点作为锚点
○ 步骤4. 调整粒度:如果原型之间的距离很近,则将它们合并或使用分层结构来调整距离
○ 步骤 5. 通过搜索每个锚点的最近位置并识别标记基因来形成原型群落
○ 步骤 6. 如果给出了特征基因集,则将原型标记基因添加到现有基因集并重新计算锚点。
○ 在此过程中,使用稳定的婚姻匹配算法将每个原型与最相似的签名进行匹配。
⑷ 类别4. 其他聚类算法
① 基于SNN(共享最近邻)模块化优化的聚类 算法
②莱顿聚类
③ 鲁汶聚类
④均值漂移聚类
⑤ DBSCAN(基于密度的噪声应用空间聚类)> ⑥谱聚类
⑦ 高斯混合
⑧【分水岭算法】(https://opencv-python.readthedocs.io/en/latest/doc/27.imageWaterShed/imageWaterShed.html)
⑨ 阈值法
⑩MST(最小生成树)
⑪曲线演变
⑫ 稀疏邻接图
⑬ SC3
⑭ SIMLR
⑮FICT
⑯模糊聚类
8. Common 2. 差异表达基因 (DEG) 分析
⑴ 定义:寻找实验组和对照组之间差异基因的过程。
⑵ DEG 的标准:根据实验设计略有不同。
①实验设计
图 22. 设计矩阵
① FC(倍数变化)
○ 定义:处理组平均基因表达量与对照组平均基因表达量的比率。
○ 问题1. 除以零:有此类问题的基因要么被预先删除,要么通过单独的解释分配特殊值。
○ 问题 2. 1 左右的不对称值:通过使用日志折叠更改解决。
② 调整后的 p 值
○ 多重测试问题:进行多重统计测试的行为本身可能会导致不准确的结论。
○ 类型 1. 系列错误率 (FWER) 的控制
○ 定义:在所有测试的假设中得出至少一个错误结论的概率。例如,FWER 为 5% 意味着在多个假设检验中即使得出一个错误结论的可能性也低于 5%。这种方法非常保守,几乎不允许误报。
○ 1-1. Sidak 校正:调整 alpha 阈值而不是 p 值,在 p 值独立时使用。
○ 1-2. Bonferroni Correction:直接调整每个 p 值,即使 p 值不独立也可以应用。这是高度保守的。
○ 类型 2. 错误发现率 (FDR) 控制
○ 定义:将实际拒绝的假设中包含原假设的概率(FDR,第一类错误率)控制在一定水平以下的方法。
○ 2-1. Benjamini–Hochberg (B&H):当检验之间的相关性相对简单时使用。
○ 2-2. Benjamini–Yekutieli (B&Y):当检验之间的相关性很复杂时使用。
○ 类型 3. FWER 和 FDR 控制可以同时应用。
③ 可视化1. 火山图
○ x 轴:记录折叠变化
○ y 轴:对数调整 p 值
○ 对于可视化满足 DEG 条件的基因分布很有用。
④ 可视化 2. MA 图
○ x 轴:对数标准化计数的平均值
○ y 轴:记录折叠变化
广泛应用于微阵列分析,适用于RNA-seq。
⑶ 统计技术
①t检验
○ 常用的统计技术之一
○ 参数统计估计
○ 取表达对数后应用 t 检验 (FPKM/TPM)
○ 由于方差估计困难,不建议用于小样本:在这种情况下,建议使用 DESeq、EdgeR、limma
○ 建议严格应用 FC 阈值
② Mann-Whitney-Wilcoxon(Wilcoxon 秩和检验)
○ 非参数统计估计
○ 不建议样本量小于 10
○ 对于小样本,建议使用limma、edgeR、DESeq2等。
○ 用于多水平单因素DEG分析» ○ 样本量小于10时,建议使用DESeq2、edgeR、limma的多级单因子模式
④ FC截止
○ 倍数变化截止值隐式假设变化恒定,例如从之前。
⑷ DEG 工具
○ 输入:原始计数
○ 假设我们可以获得更好的基因方差估计,而不假设它们都具有相同的方差。
○ 不支持随机效应建模或混合效应建模:limma有些灵活,所以可以做。
○ 基于【负二项式模型】(https://jb243.github.io/pages/1626#7-negative-binomial-distribution-)
○ 负二项式模型可能对异常值敏感:最新版本的 DESeq2 添加了额外的功能,以对异常值具有鲁棒性。
○ 在以下两个选项下使用经验贝叶斯模型估计方差:
○ 选项 1. 分散
○ 步骤1. 对每个基因进行最大似然估计(MLE)以获得图中的黑色数据点。
○ 步骤 2. 从黑点导出红色趋势线,作为先验平均值。
○ 步骤 3. 计算 MAP(最大后验)估计,如蓝色箭头所示。
○ 步骤 4. 未向先验收缩的数据点可被视为异常值,用蓝色圆圈标记。
图 23. DESeq 色散模式
○ 选项 2. 折叠更改(可选)
○ DESeq2 为 log2FC 估计提供收缩(旧版本将此设置为默认值):lfcShrink()
○ 所有基因的 LFC 首先用作先验来估计趋势,并“缩小”LFC 似然估计。
○ 示例:在下图中,您可以看到绿色数据点,这些数据的可能性很高,但没有很好地向先验收缩。
图 24. DESeq 折叠更改模式
黑线:之前 实线:未缩小的估计 虚线:缩小的 LFC 预估
○ 使用库克距离标记每个基因的异常值:距离是衡量最小二乘分析中数据点影响力的指标。
○ 样本量小:从分析中删除基因。
○ 大样本量:如果检测到特定基因的异常值样本,请将这些样本从分析中排除。
○ 注意获取差异表达基因和可视化的不同预处理方法。
② 边缘R
○edgeR(精确)、edgeR(GLM)
○ 使用 TMM 归一化和负二项式广义线性模型 (GLM)。
○ EdgeR 默认情况下会过滤掉总读取数 <5 的基因,以估计常见的离散值。
○ 估计离散度的几种选择
○ 通用:使用单一、通用的色散估计。不推荐。
○ 趋势:根据趋势函数估计。
○ Tagwise:贝叶斯调节方法,类似于 DESeq2。
③ 边缘R-QLF
○ 将方差建模为 Var(ygi) = σg2(μgi + μgi2φ) 以允许过度离散。» ○ 离差 φ:基因丰度 (φ(A)) 的函数,它适合经验均值-方差关系的趋势估计的离差。
○ σg2 是基因特异性方差(准离散参数)。这与经验贝叶斯方法相符。趋势函数在基因丰度和原始 QL 离散度估计之间拟合。然后,使用经验贝叶斯方法,原始 QL 估计(可能性)缩小到该丰度的平均拟合趋势。
○ μgi + μgi2φ(即 σg2 = 1)对应于标准负二项式的方差。
④ 极限
○ 输入:原始计数(避免使用标准化数据,如带有 voom 的 FPKM)
○ 第一第一。将读取计数转换为日志 CPM。
○ 第二第二。使用经验贝叶斯方法(eBayes();假设正态分布):消除计数数据中存在的均值和方差之间的相关性。
○ 第三第。基于广义线性模型(GLM)的DEG探索。
○ 用于获取 DEG 的矩阵也用于可视化
○ 假设我们可以获得更好的基因方差估计,而不假设它们都具有相同的方差。
○ 一般来说比edgeR和DESeq2更保守。
○ 输入:FPKM(当原始计数数据不可用时有用)
○ 第一第一。将 FPKM 转换为 log2 刻度
○ 第二第二。使用 trend = TRUE 选项运行 limma 的 eBayes() 函数
○ 此方法类似于微阵列分析中使用的方法,也类似于 limma-trend 方法
⑤ 序列
⑥ CuffDiff:使用 FPKM 值。
⑦ BaySeq
⑧ EBSeq
⑨ DEGSeq
⑩ NOISeq
⑪ 泊松序列
⑫ SAMSeq
⑬ NetBID2
⑭ scVI
○ 提供消除批次效应的标准化表达式,并提供基于它们计算 DEG 的代码(参考)。
○ 提供的用于计算 DEG(差异表达基因)的代码不仅速度慢而且不准确:从 10 个数据集中删除批次效应可能与仅在其中 2 个数据集中删除批次效应略有不同,这有时会导致完全相反的 DEG 模式。
○ 建议使用scVI标准化表达式和
scanpy.tl.rank_genes_groups来识别DEG。
⑮ 无聚类DEG算法
○ 基于分布测试:SingleCellHayStack、MarcoPolo(开/关方法+泊松分布假设)、SEMITONES、SchayStack
○ 基于差异统计:DUBStepR、M3Drop、Cofea、BigSur
○ TAMER:采用互斥性(≠反相关)。
○ GSPA:将邻接矩阵乘法(=随机游走)应用于小波分析
⑸ 技术选择
| 样本量 | 计数深度 | |||
|---|---|---|---|---|
| 低计数(~20 M 或更少) | 高计数 (~30 M+) | |||
| 小 (3-9) | 基于贝叶斯计数的测试(例如,edgeR QLF、DESeq2) | 贝叶斯方法(基于计数或连续) | ||
| 中 (10-30) | 基于贝叶斯计数的测试(例如,edgeR QLF、DESeq2) | 基于计数或连续;可能是非参数的 | ||
| 大 (>30) | 基于计数的测试 | 许多选项:基于计数、连续、非参数 |
表 7. 技术选择
9。 Common 3. 基因集富集
⑴ 概述
① 定义
○ 将基因组作为一个整体进行分析,而不是单个基因。
○ 【基因评分】(https://jb243.github.io/pages/2215):指基因列表生成的值。
○ 签名:由基因名称、FC(倍数变化)值或 p 值组成的数据框。 基因评分也可以被视为签名。
② 方法
○ 方法1. ORA(过度代表性分析)
○ 测试特定路径是否包含比偶然预期更多的 DEG。
○ 归类为竞争性测试而不是独立测试。
○ 独立测试:仅使用感兴趣的基因集评估差异表达或特定模式。
○ 竞争性测试:将感兴趣的基因集与整个背景基因集进行比较,以确定其是否包含大量与特定途径相关的基因。
○ 1-1. Fisher 精确检验(超几何检验)
表 8. 基因集分析中的列联表
○ 统计 1. 概率:与样本相似的概率。
○ 统计数据 2. 优势比:显示 GO 和基因集是否相似或不相似。
○ 统计3. 基因比例:表示A/(A+B),即常见基因与输入基因集的比例。
○ 统计4. 计数:通常表示A,两个集合之间的交集元素的数量。
○ 代表性过高分析的示例。
○ 1-2: 修改的 Fisher 精确检验(例如 DAVID)
图 25. 改进的 Fisher 精确检验
○ 当 DEG 数量较少时,影响显着减小,而当数量较多时,影响很小。
○ 代表性过高分析的示例。
○ 方法2. 基因集排序(评分)方法
○ 使用跨基因的排名或显着性值测试每个路径。
○ 示例:GSEA、singscore
○ 方法3. 基于网络的方法
③ 解释
○ 根据实验组和对照组之间的表达对基因进行排序,然后计算富集分数。
○ 如果查询基因集中的基因高度集中在排序基因列表的两个极端(例如前 10 名或后 10 名),则富集分数会增加。
① 概述
○ 基于非参数排列。» ○ 广泛使用,特别是与人类和癌症相关的数据。
② 输入
○ 类型 1. 表达式文件(.gct 或 .txt):表达式值
○ 类型 2. 表型标签 (.cls):组名称(例如,对照、正常)
○ 类型 3. 基因注释 (.chip):基因名称
○ 类型 4. 基因集数据:基因集
○ 类别 1. M1H:小鼠直系同源标志基因集等。
○ 类别2. M1:位置基因集等。
○ 类别 3. M2:策划的基因集等。
○ 类别 4. M3:监管目标集等。
○ 类别5. M5:本体基因集等。
○ 类别6. M8:细胞类型特征基因集等。
③实施
○ GSEA_R:R 包。将迭代次数增加到 10,000。更适合大样本量的实验。
○ R 中的一些“gsea”实现(例如 fgsea)是不同的。
④ 输出
○ FDR(错误发现率)
○ NES(标准化富集分数)
○ 前沿子集
⑤Python代码
受保护_33
⑶ 类型2. GO(基因本体)
① 整合所有物种基因和基因产物表达的重大生物信息学举措
②按照CC(细胞成分)、MF(分子功能)、BP(生物过程)等分组。
③ 证据代码:利用各种信息源将基因分配给基因本体术语。信息来源存储在证据代码中。
○ 类型 1. 实验证据代码:实验 (EXP)、直接测定 (IDA)、物理相互作用 (IPI)、突变表型 (IMP)、遗传相互作用 (IGI)、表达模式 (IEP)
○ 类型 2. 计算证据代码:序列或结构相似性 (ISS)、序列同源性 (ISO)、序列 (ISA)、序列模型 (ISM)、基因组背景 (IGC)、审查计算分析 (RCA)
○ 类型 3. 作者声明:可追踪作者声明 (TAS)、不可追踪作者声明 (NAS)
○ 除一种类型的证据代码外,所有类型的证据代码均由策展人分配;唯一的例外是从电子注释 (IEA) 中推断出来的。
③实现:通过[R]实现的功能(https://jb243.github.io/pages/2156)
受保护_34
④ 结果示例
图 26. GO 分析结果示例
○ Count: 输入基因集与每个GO term之间交集的大小,即共同基因的数量
○ p.调整: 使用 Fisher 精确检验和 Benjamini-Hochberg (B&H) 调整输入基因集和每个 GO 术语之间的 p 值
○ GeneRatio: 基因比例,即输入基因集中常见基因的比例(ref)
○ ▶ 如何解读 GO 图
⑤ 网站
⑷ 类型3. DAVID(功能注释生物信息学微阵列分析)
① 提供提交基因的生物学解释:尽管是为微阵列数据开发的,但仍然效果相对较好
② 用于基因表达数据或基因组区域限制在 TSS < 1-5 kb 范围内。
⑸ 类型4. MSigDB(分子特征数据库)
①分类
○ H:Hallmark 基因集(50 个术语)
○ C1:位置基因集(299 个术语)
○ C2:精选基因集(6226 个术语)
○ C3:调控目标基因集(3556个术语)
○ C4:计算基因集(858项)» ○ C5:本体基因集(14765个术语)
○ C6:致癌特征基因集(189 个术语)
○ C7:免疫特征基因集(4872 个术语)
○ C8:细胞类型特征基因集(302 个术语)
② GO 和 MSigDB 的区别
○ GO:多种
○ MSigDB:以人为本。一些老鼠(可以转换成老鼠)
⑹ 类型 5. EnrichR
① 使用各种基因集信息进行基因集分析。
② richr(R 包;基于在线的 API)或网络工具
③使用Fisher精确检验和Benjamini-Hochberg (B&H)调整进行统计显着性计算。
④ 显示提交的基因和其他注释基因集之间的一致性
⑺ 类型6. ToppGene, ToppFun等:网络工具
① 转录组
② 蛋白质组
③ 调控组(例如,TFBS、miRNA)
④ 本体(例如GO、Pathway)
⑤ 表型(例如人类疾病、小鼠表型)
⑥ Pharmacome(例如药物-基因协会)
⑦ 文献共引
⑻ 类型 7. iLINCS:药物数据库
⑼ 类型 8. g:Profiler
⑽ 类型9. KEGG(京都基因和基因组百科全书)
① 日本创建生化通路数据库(1995)
② 生物学领域被引用最多的数据库之一
③ 包括以生化途径为中心的系统、基因、健康、化学等相关的各种信息。 577 途径。
④ 方法:pathview Bioconductor 软件包,https://pathview.uncc.edu/, g:Profiler(基于 Web 的工具)
⑤ KEGG网站允许您对基因进行着色以显示差异表达。
⑾ 类型 10. 信任
⑿ 11 型. 代谢途径
⒀ 类型 12. REAC(反应组)、WP、TF 等。
① 显示与提交基因相关的通路
② 基于Web的工具:g:Profiler、iLINCS
⒁ 类型 13. IPA(独创性路径分析)
① 商业软件
② 通过组学数据可视化通路和网络
③ 可以利用数据识别因果机制和关键因素
④ 包括规范路径、上游监管分析和更新信息等信息
⒂ 类型 14. SPIA(信号通路影响分析)
① 显示与提交基因相关的信号通路拓扑
⒃ 15 型. MGI
⒄ 类型 16. WP:可以使用基于网络的工具进行检查,例如 g:Profiler
⒅ 类型 17. TF:可以使用基于 Web 的工具进行检查,例如 g:Profiler
⒆ 类型 18. GSVA
⒇ 19 型. AUCell 封装
⒇ 类型 20. HOMER 基序分析
⒇ 21 型. 金标准方法
⒇ 22型. Ensembl基因
⒇ 类型 23. UCSC 已知基因
⒇ 类型 24. ssGSEA(单样本 GSEA):ssGSEA 为每个样本生成富集分数。然后,ssGSEA 结果可用于查找数据集中的异常值或比较样本之间的富集路径。
⒇ 类型 25. singscore R 包:与 ssGSEA 类似。它为每个样本生成富集分数。然后,结果可用于查找数据集中的异常值或比较样本之间的富集路径。
⒇ 类型 26. LRpath:Web 工具。逻辑回归
⒇ 27型. 黑豹:与通路相关。
⒇ 类型 28. Biocarta:与途径相关。
⒇ 类型 29. MeSH 术语:与疾病相关。
⒇ 类型 30. DisGeNET:与疾病相关。
⒇ 31 型. 细胞带
⒇ 类型 32. Babelomics:网络工具⒇ 类型 33. clusterProfiler:R 包
⒇ 类型 34. goseq:R 包。也用于空间转录组学的 GSE 分析。
⒇ 类型 35. GOrilla:非参数基于排名。
⒇ 类型 36. RNA 富集:逻辑回归。 LRpath网站
⒇ 类型 37. chip-enrich (示例数据,R code):ChIP-seq 的 GSE 分析。对于具有尖锐峰值的数据集。
⒇ 类型 38. Broad-Enrich:专为组蛋白数据分析而设计。也包含在chipenrich 包中。适用于广泛的基因组区域。
⒇ 类型 39. GREAT(基因组区域注释工具丰富):利用二项式分布和 Fisher 精确检验,减少误报。
⒇ 类型 40. iDEA:scRNA-seq 的 GSE 分析。
⒇ 类型 41. Giotto:空间转录组学的 GSE 分析。对每个点使用超几何测试、PAGE 或基于等级的测试。
⒇ 类型 42. SPATA2:执行 hypeR 包,该包使用超几何测试。也可应用于空间转录组学。
⒇ 类型 43. GIGSEA:GWAS 的 GSE 分析
⒇ 类型 44. MAGMA:GWAS 的 GSE 分析
⒇ 类型 45. i-GSE4GWAS:GWAS 的 GSE 分析
⒇ Missmethyl 包中的类型 46. gometh 函数:CpG 位点的 GSE 分析
⒇ 类型 47. 相机(相关调整平均等级分析)
⒇ 类型 48. Seq2pathway:R/Bioconductor 包
⒇ 类型 49. 很棒(基因组区域富集注释工具):具有非常夸大的 1 型错误。
⒇ 50 型. 电子实用程序
⒇ 类型 51. AgentAPI
⒇ 52 型. HPO
⒇ 类型 52. 维基路径
⒇ 类型 53. PubMed
⒇ 54 型。 生物星球
⒇ 55 型. CDD
⒇ 类型 56. BLAST-KNN:基于序列。利用序列相似性搜索。
⒇ 类型 57. PFresGO:基于序列和 GO 术语。利用 PLM 和多头注意力。
⒇ 58 型. Goretriever:基于文献。利用法学硕士和学习排名。
⒇ 类型 59. 辅因子:基于序列、PPI 和结构。利用相似性搜索和集成。
⒇ 类型 60. StarFunc:基于序列、PPI、结构和 Pfam。利用 PLM、相似性搜索和随机森林。
⒇ 类型 61. DeepGOPlus:基于序列。利用 CNN 和相似性搜索。
⒇ 类型 62. InterLabelGO+:基于序列。利用 PLM、标签相关性学习和相似性搜索。
⒇ 类型 63. NetGO3.0:基于序列、PPI、InterPro、文献。利用逻辑回归和学习排序。
10。 Common 4. 基因相互作用分析
⑴ 类型1. 【细胞与细胞相互作用】(https://jb243.github.io/pages/841)(CCI,配体-受体相互作用)
①原理:当一个细胞配体高表达,另一个细胞受体高表达时,两个细胞中配体和受体相互作用
② 基于bulk-RNA-seq
○ 鱿鱼
○ IPA(上游监管者独创路径分析)
○ Omnipath:请参阅此处,获取输出包含特定基因的所有配体-受体对的代码。
○ 生物密码
○ 前查找器
③基于scRNA-seq
○ CellTalkDB (人类数据库文件, 鼠标数据库文件)» ○ CellPhoneDB (教程)
○ ICELLNET
○ NicheNet
○ 软体SC
○ 细胞语
○ sc张量
○ CCC探索者
○ 连接体
○ 拉米洛夫斯基
○ FlowSig:图形因果建模(完整的部分有向无环图;CPDAG),条件独立性测试
○ scSeqComm:来自 Reactome + TTRUST + RegNetwork 的 LR 对
○ 南美
○ SingleCellSignalR
○ SCITD
○ MOFA蜂窝
○ MOFAtalk
○ 多利基网络
○ 张量-cell2cell
④ ST为主
○ 乔托
○ 斯帕塔2
○ CellPhoneDB v3
○ 学习
○ SVCA
○ 迷雾
○ NCEM
○ STopover:采用扩散和 Jaccard 索引
○ 细胞信号
○ 水疗谈话
○ stMLnet
○ 全息网
○ 深林公司
○ GCNG
○ conST
○ 七巧板
○ 对话
○ 细胞迷航
○ LIANA+:一个集成了 CellphoneDB、CellChat、Connectome、scSeqComm、NATMI、Moran、PyDESeq2、Tensor-cell2cell(高阶分解)、scHOT、SpatialDM 等的一体化框架。
○ 空间
○ 迷雾R
○ 斯帕OTsc
⑤ 蛋白质-蛋白质相互作用(PPI,分子对接)
○ AlphaFold2 多聚体、AFM-LIS、AlphaFold3
○ Boltz-2
○ 深度DTA
○ 深度DTAF
○ DeepFusionDTA
○ 图DTA
○ CAPLA
○ 吉娜
○ 斯米娜
○ 滑行
○ EquiBind
○ 坦克绑定
○ DIFFDOCK
○ STRINGdb
⑵ 类型2. 网络分析:全基因关联研究(GWAS)、蛋白质-蛋白质相互作用(PPI)等。
① 2-1. 生物网络构建
○ 1型. 基因调控网络
○ 类型 2. 蛋白质-蛋白质相互作用网络
○ 类型 3. 共表达网络
○ 基于生物相互作用或基因表达数据构建网络是可能的。
○ 构建网络后,可以使用各种网络测量/指标来识别生物学上重要的基因。
② 2-2. 监管网络分析
○ 使用类似套索的方程,例如
min |y - βX| + λ|β|。
○ 一种计算蛋白质-蛋白质网络、基因表达网络和 TF-目标基因网络中边缘一致性的算法。
○ 通过更新 TF 和目标基因之间的调控网络边缘来构建网络。» ○ 根据基因-基因相互作用识别群体差异。
○ 基因表达调控机制
○ 转录起始频率的调节
○ 转录延伸的调节
○ 替代转录起始 (ATI)
○ 选择性剪接 (AS)
○ 替代聚腺苷酸化 (APA)
○ RNA 干扰 (RNAi) 导致的 RNA 降解
○ 长链非编码 RNA (lncRNA) 的 RNAi 干扰
○ 翻译启动规定
○ 染色体重塑和表观遗传调控
③ 2-3. 特定样本网络
○ 提出为单个样本构建生物网络的方法,而不是使用所有样本的数据或构建基于群体的网络。
○ 通过此,可以发现个体患者中受特定基因影响的基因差异。
④ 2-4. 模块/社区检测
○ 假设由生物成分组成的网络不是随机的,而是由执行特定功能的模块组成。
○ 假设生物网络内的节点(例如基因)形成群落(例如路径)。
○ 可以执行社区/模块检测,一种网络分析技术。
○ 检测到的模块/群落可以通过 GSEA 分析进行生物学解释。
⑤ 2-5. Hub基因检测
○ 网络类型:度中心性、介数中心性、紧密度中心性、特征向量中心性、参与系数、Pagerank
○ 通过各种指标提取模块/社区内的核心基因,以寻找网络分析技术中的枢纽
⑥ 示例 1. ToppNet:识别网络中候选基因的相对重要性。
⑦ 示例 2. ToppGenet:对蛋白质-蛋白质相互作用网络中的邻近基因进行排名。
⑧ 示例 3. GeneMANIA:使用基因组学和蛋白质组学数据显示功能相似的基因。
⑨ 示例 4. SCINET:使用 scRNA-seq 算法。
⑩ 实施例5. MEAGA(基于最小距离的遗传关联富集分析):利用疾病相关功能/通路中易感位点的基因在生物相互作用组中彼此更接近的事实。
⑪ 示例6. X2K:显示上游监管网络。
⑫ 示例 7. WGCNA:基于 TOM 的聚类。
⑬ 示例 8. Louvain / Leiden 算法:基于模块化的聚类
⑭ 实施例9. IPA(Ingenuity Pathway Analysis):独创性通路分析的上游调控因子分析
⑮ 示例 10. CCCExplorer
⑯ 实施例 11. 连接体
⑰ 示例 12. 鱿鱼
⑱ 示例 13. spata2
⑲ 示例 14. ALIGATOR:分析与 GWAS 相关的基因是否在特定途径中过多表达。目标基因集(GO、KEGG)。确保唯一性并应用多重测试校正。
⑳ 示例 15. INRICH:分析基因集是否过度聚集在特定位置。使用基于排列的测试。
㉑ 实施例16. DAPPLE:分析基因在PPI网络内是否过度连接。评估实际网络与随机网络的连通性。
㉒ 示例 17. PiNET:评估疾病相关基因在蛋白质-蛋白质相互作用 (PPI) 网络中是否过度连接。它对肽部分进行注释、绘图和分析。
⑶ 类型3. TF(转录因子)分析
① 概述» ○ 真核生物中的RNA聚合酶不能单独与启动子结合。
○ 当一般转录因子和特异性转录因子与基因上游的各种调控序列(如TATA、CAAT)结合时,RNA聚合酶即可启动转录。
○ 转录因子的数量约为1,600个。
○ 先锋因子:一种特殊的转录因子,即使染色质处于关闭状态也能结合。它是第一个结合的转录因子,但不太常见。
○ CTCF 转录因子:与其他 TF 相比,与 DNA 结合的时间更长(约几分钟)。
② 算法
○ 无花果
○ scGRNom
○ 远程
○ 三角测量
○ 风景区
○ 深度扫描电镜
○ 推断器
○ Sc-compReg
○ 比特福姆
○ 多萝西娅
11。 Common 5. 细胞类型映射分析
⑴【一般细胞类型标注】(https://jb243.github.io/pages/1782)
① 目的
○ 意义1. scRNA-seq中的细胞类型分析和ST中ROI的深入分析可以有效减轻样本选择偏差造成的批次效应。
○ 意义2. 基于细胞类型而不是基因表达分析进行分析使结果更容易理解。
② 方法1. 基于聚类
○ 定义:对 scRNA-seq 数据进行聚类,然后根据每个聚类内基因的差异表达来标记细胞类型。
○ 缺点1. 按分辨率参数划分:同一簇中的所有细胞不一定属于同一细胞类型。
○ 缺点2. 相同类型的细胞可能根据其状态被分成不同的簇。
○ 缺点 3. 比较不同平台上的细胞类型标签具有挑战性(例如,免疫细胞、DC 与 cDC)
③ 方法2. 元标记
○ 定义:根据分数或多轮聚类将多种细胞类型附加到一个细胞。
○ 示例:将单个细胞同时标记为免疫细胞、DC 和 cDC(例如 xCell)。
④ 方法3. ► 单细胞基础模型方法
○ 定义:使用语言模型创建用于指定细胞类型的单个标签器。
○ 基于 BERT:使用基于 Transformer 编码器的架构。
○ 采用预训练 → 微调方法。
○ 零射击能力实际上毫无用处。
○ Geneformer-V2-316M、Geneformer-V2-104M、Geneformer-V2-10M 等
○ scGPT
○ 基于 GPT:使用基于 Transformer 解码器的架构。
○ 采用预训练 → 微调方法。
○ 预训练模型的零样本性能也相当出色。
○ 全人模型等
○ GenePT 和 scGenePT
○ UCE(通用细胞嵌入):多物种数据集
○ 概述
○ 103种组织类型,品种多
○ 输入是原始 scRNA-seq 计数矩阵和从 ESM2 获得的蛋白质嵌入
○ ESM2也用于SATURN算法:跨物种scRNA-seq整合
○ 4层默认模型和6.5亿参数的33层模型
○ 元数据不影响嵌入
○ 与 TranscriptFormer 的共同点
○ 对于非零计数,基因及其表达值用作细胞的基因表达表示»» ○ 通过 ESM-2 模型的氨基酸嵌入来嵌入蛋白质编码基因
○ 如果多个蛋白质与单个基因匹配,则取其 ESM-2 蛋白质嵌入的平均值
○ 与 TranscriptFormer 的差异
○ 为了构建尽可能通用和简单的表示,UCE 仅将输入限制为表达式数据(计数):不合并元数据
○ 也利用染色体位置信息。
○ scFoundation:仅限人类
○ Cell2Sentence (C2S), C2S-Scale
○ 使用高表达基因列表(通常 100 个)作为模型嵌入
○ 使用 NWP(下一个单词预测)方法微调 GPT-2 等生成语言模型
○ 使用
predict_cell_types_of_data函数预测细胞类型。
○ 类型 1.
vandijklab/pythia-160m-c2s:快速。
○ 类型 2.
vandijklab/C2S-Pythia-410m-cell-type-prediction:更强。
○ 类型 3.
vandijklab/C2S-Scale-Pythia-1b-pt:大。介绍多种应用,例如 C2S 规模的空间转录组学。
○ CELLama
○ scBERT
○ 利基代理
○ 协奏曲
○ 传输
○ GeneCompass: 人/小鼠
○ 甘油三酯
○ 细胞LM
○ CellPLM
○ scMulan:仅限人类
○ 向语言模型提供诸如
(Heart, 0), (Cardiomyocyte cell, 0), (A2M, 0.2), (ZNF385B, 0.6), …, (CD83, 0.1), (E, 0)之类的输入。
○ 使用所有非零表达的基因作为输入。
○ 利基形态:人类/小鼠
○ scPRINT:人/小鼠
○ 泰迪
○ Xu et al.: 多物种数据集
○ TranscriptFormer:使用 1.12 亿个细胞进行训练。麻省理工学院许可证
○ 概述
○ 1.12 亿个细胞,跨越 15.3 亿年
○ 类型:TF-Metazoa(所有 12 个物种)、TF-Exemplar(人类和 4 个模式生物)和 TF-Sapiens(仅限人类数据;5700 万个细胞)
○ 麻省理工学院许可证
○ 与 UCE 的共同点
○ 对于非零计数,基因及其表达值用作细胞的基因表达表示。
○ 使用 ESM2 模型通过氨基酸嵌入来嵌入蛋白质编码基因。
○ 如果多个蛋白质与单个基因匹配,则取其 ESM2 蛋白质嵌入的平均值。
○ 与UCE的区别
○ TranscriptFormer 可以将元数据合并到嵌入中(CGE:上下文化基因嵌入)。
○ 由于 TranscriptFormer 的标准推理代码仍处于研究原型水平且难以使用,因此出于实用/基准目的,增强的包装器至关重要,包括:
○ config.json 与后备值的安全合并
○ 自动补充缺失的 aux_cols
○ 自动化处理重复基因/NaN
○ 内置端到端分类器
○ 概述
○ 由 GeneBIO AI 开发,由 Chan Zuckerberg Initiative (CZI) 主办。
○ 采用基于 BERT 的 MLM(掩码语言模型)方式进行训练来预测基因表达。
○ AIDO.Cell系列共有4个型号:3M、10M、100M、650M
○ 可以使用
extract_features.py中的model(model.transform(...))函数获得嵌入。
○ 限制
○ 元数据不影响嵌入。
○ Flash-attn 后备问题:与嵌入无关,因此可以使用 PyTorch SDPA 代替嵌入。»» ○ BATCH_SIZE 较大时会出现 GPU 内存问题。
○ scConcept:采用对比学习进行模型训练,而不是掩码语言建模(MLM)
○ 细胞爱马仕
⑵ Bulk RNA-seq
① xCell:基于R。定义每个人类基因的权重。
② immunedeconv:基于R的基准测试算法。
○ 对于人类数据:可用模式包括
quantiseq、timer、cibersort、cibersort_abs、mcp_counter、xcell、epic、abis、consensus_tme和__受保护_149__。
○ 对于鼠标数据:可用模式包括
mmcp_counter、seqimmucc、dcq和base。
③BayesPrism:贝叶斯算法。需要 scRNA-seq 参考。
④ MixupVI
○ 原样使用 scVI 架构。
○ 在变分推理中使用的ELBO中添加了Mixup loss,使得潜在空间具有线性。
○ 为了进行基准测试,不仅提供 MixupVI,还提供 OLS、NNLS、DWLS、Tape、Scaden、NuSVR (CIBERSORT)、RLR 和 WNNLS。
⑶ scRNA-seq(单细胞RNA测序)
○ Seurat:基于 R。采用一种称为“标签传输”的方法。
○ scanpy:基于Python,尤其与通过摄取进行细胞类型分析相关。
○ Scanorama:基于Python。
○ celltypist 和 celltypist2:基于 Python。逻辑回归模型。将预定义的引用保存在
.pkl文件中。
○ SingleR:自动化细胞类型注释工具。基于参考。
○ scPred:自动化细胞类型注释工具
○ CellAssign:概率模型
○ 萨金特:基于评分的方法
○ scTab、SELINA、Spoint、Tangram、TACCO、InsituType、Symphony、CHETAH、scibet、scmap、CellID、sccatch、SCINA、scsorter、CellMarker 2.0、Signac、ArchR、cisTopic、snapATAC、 MARS、scArches、treeArches、CellHint
⑷ ST(空间转录组学)> ○ CellDART、spSeudoMap、RCTD(基于R)、MIA分析:(包括两者) 富集 和耗尽 分析)、Cell2location、 SPOTlight、DSTG、CellTrek (共同嵌入 scRNA-seq 和 ST 后,它使用基于距离的图和随机森林执行细胞类型标记)、CytoSpace、Tangram、BayesPrism、DestVI、 立体镜、SpatialDWLS、 GIST、GraphST、TACCO、 平滑、CARD、 Cellscope、Starfysh、Seurat、AdRoit、Spatial-ID(来自 scRNA-seq 的注释转移)、 SpaTM-G、SpaOTsc、NLSDeconv、Redeconve、SpaCET、SpatialPrompt、SPADE、SpatialDecon、SONAR、AntiSplodge、UniCell Deconvolve、SpatialDDLS、TransformerST、DEEPsc、 RedeHist、LETSmix、scResolve、SPACEL-Spoint、STEM、SpaDecon、Bulk2space、SpatialcoGCN、SD2、GNNDeconvolver、GTAD、STdGCN、STRIDE、SMART、stVAE、POLARIS、STIE、MUSTANG、BayesTME、RETROFIT、SpatialScope、ST-assign、MAST-Decon、EnDecon、Cottrazm、 CellsFromSpace、FAST、CellPie、SpiceMix、LANTSA、NovoSparc、scDOT、DOT、SpaTrio、LANSTA、SDDLS、BANKSY、MERINGUE、 SpaGCN、BayesSpace、FICT、CMAP、 SpatialZoomer、Spotiphy(接受 scRNA-seq 和 ST 的输入)、DistMap、SpaGE
12。 Advanced 1. 选择性剪接分析(AS 分析)
⑴ 概述
① AS 分析可以使用现有数据和剪接感知对准器进行。
② 然而,随着被选为2022年度技术的长读长测序的出现,更准确的分析成为可能。
⑵ 长读长测序
① 与短读长测序相比,测序间隙更少。
图 27. 长读长测序与短读长测序
② 优点 1. AS 分析:能够识别可变剪接事件和亚型。
③ 优点2. 促进表观遗传学和转录组学的整合。
④ 示例 1. Pacific Biosciences SMRT(单分子实时)测序:平均读长约为 20 kb。
⑤ 实施例 2. 牛津纳米孔测序:平均读长约为 100 kb。
⑶(参考)选择性剪接事件
① SE(跳过外显子):包含或排除整个特定外显子。> ② A5SS(替代 5’ 或 3’ 剪接位点):外显子 5’ 或 3’ 剪接点的不同用法,而不是整个外显子。
③ MXE(互斥外显子):当另一个外显子剪接时,互斥外显子被剪接,反之亦然。
④ RI(Retained Intron):不编码氨基酸的内含子被保留或剪接。
⑷(参考)外显子测序:通常与基因测序相对照。
① 外显子符号示例:
○ chr15:63553600-63553679:-
○ chr15:56967876-56968046:-
○ chr7:7601136-7601288:+
○ chr11:220452-220552:-
② 基因符号示例:
○ 人类基因:SIRPA、HBB-BS等
○ 小鼠基因:Sirpa、Hbb-bs 等
⑸ 类型 1. 基于事件的 AS 定量:使用基于计数的模型划分为外显子进行定量。
① PSI 值:量化每个外显子事件的 PSI。
○ 使用百分比剪接 (PSI) 值量化 AS 事件。
○ PSI值=包含读数/(包含读数+排除读数)
○ 代表性工具:rMATs
② 外显子使用情况:根据外显子计数进行分析。
○ 根据映射区域,Reads可以分为外显子reads和连接reads。
○ 外显子读取:外显子区域内的映射读取。
○ 连接读取:剪接连接处的映射读取。
○ 外显子读取按每个外显子进行计数,以进行外显子使用(外显子级表达)计算。
○ 代表工具:DEXseq
⑹ 类型 2. 基于异构体的 AS 定量
① 定义:使用统计模型在转录本水平估计每个亚型转录本的表达。
② 目的:如果可以定义目标亚型,则可以提高约束、诊断等方面的功效。
③ 代表工具:RSEM
④ 用于异构体探索的数据库
○ UniProt:最著名的蛋白质相关数据库。
○ Ensembl
○ Reactome
○ GPP 门户网站
○ NCBI 程序集(示例):执行针对 FASTA 和 GTF 文件 的代码(例如、pyfaidx)
13。 Advanced 2. 轨迹分析
⑴ 概述
①CNV(拷贝数变异):细胞分裂异常引起的染色体数量异常。指染色体缺失或非整倍体。
② SNP(单核苷酸多态性):特定核苷酸序列的差异。
③ 这只能通过直接测序方法(例如 Visium FF、scRNA-seq)实现,而不能通过基于探针的方法(例如 Visium FFPE)实现。
④ CNV分析中,p指短臂,q指长臂。
⑵ CNV分析算法
① CopywriteR:基于WGS。分析脱靶阅读深度。
② CNVkit:基于WGS。分析读取深度的偏差。
③ ASCAT:基于WGS
④银杏:基于scDNA-seq。分析读取深度的偏差。
⑤ InferCNV:基于scRNA-seq。
⑥ CopyKat:基于 scRNA-seq。
⑦ Clonalscope:基于 scRNA-seq。
⑧ CONICSmat:基于 scRNA-seq。
⑨ HoneyBADGER:基于 scRNA-seq。
⑩ CaSpER:基于 scRNA-seq。
⑪ Numbat:基于 scRNA-seq。
⑫ SpatialInferCNV:基于 ST。
⑬ SPATA:基于 ST。
⑭ STmut:基于ST。
⑮淀粉:ST为主。
⑯ CalicoST:基于 ST。
表 9. CNV 分析算法总结
⑮ InferCNV 和 CopyKat 与 Numbat 相比的问题(ref)» ○ 问:与我的其他 CNV 调用程序相比,Numbat 生成的 CNV 配置文件似乎存在全局基线偏移;具体来说,Numbat 认为在其他分析中看似中性的细分市场为收益,而对于看似亏损的细分市场则为中性。
○ 答:许多现有方法(例如 InferCNV/CopyKAT)推断相对于中位倍性的拷贝数变异,这会稀释异常区域的信号或由于超二倍体或亚二倍体引起的基线偏移而将中性区域误认为异常区域。相反,Numbat 首先尝试根据等位基因证据(平衡等位基因频率)识别二倍体区域,并使用这些区域作为 CNV 调用的基线。
⑶ SNP分析算法
① Sniffle、Sniffle2:长读DNA-seq-base
② SCmut:基于scRNA-seq。
③ scSNV:基于scRNA-seq。
④ SComatic:基于scRNA-seq。
⑤ STmut:基于ST。
⑷ 重复序列分析算法
① 概述:在基因组参考中,重复序列要么被 N 掩码,要么转换为小写。
② 实验方法
○ Southern 印迹
○ 桑格测序
③ 串联重复序列识别算法
○ 重复遮罩
○ 串联重复序列查找器
○ HipSTR:基于 Illumina 测序数据
○ ExpansionHunter:基于Illumina测序数据
○ RepeatHMM:基于长读序列的STR检测
○ STRique:基于长读信号的 STR 检测
○ DeepRepeat:将长读长测序数据的原始电信号转换为图像并应用 CNN
⑸ 轨迹分析流程
①沿伪时间的基因表达:Monocle (ref1, ref2, ref3), TSCAN([参考](https://doi.org/10.1093%2Fnar%2Fgkw430)),Slingshot([参考](https://doi.org/10.1186%2Fs12864-018-4772-0)),PAGA,scEpath,stLearn,[SpaceFlow](https://github.com/hongleir/SpaceFlow), SIRV、PHATE(基于亲和力的轨迹嵌入的热扩散潜力)、RVAgene(使用 scRNA-seq 自动编码器)、FlatVI、STORIES(使用最佳传输)、PRESCIENT
③ 轨迹谱系:tradeSeq (ref)、LinRace (ref)、SCORPIUS (用于 scRNA-seq) ([参考](https://github.com/rcannood/SCORPIUS)),CellRank
④ 不同批次样品轨迹分析:Phenopath (ref)、Condiments (ref)、Lamian ([参考](https://www.nature.com/articles/s41467-023-42841-y))
⑤ RNA 速度(剪接与未剪接):Velillary、scVelo、STARsolo、dynamo、MultiVelo ([参考](https://www.nature.com/articles/s41587-022-01476-y.epdf?sharing_token=M2W6sq3MAWKunZfbSAytONRgN0jAjWel9jnR3ZoTv0OdGj1T8B 8MFii0PR-J_XYmuYFJPW-ydKHESAJwtpQYdR1tFvy_dcsDI4ppX8nSdJL5UwvaNk5U–STVGpJ48A8gQyjyqY1EJgOJykui6AJ55Qoo3IPqLjsw6Dbez0nJ3Q%3D)), VeloVAE、cellDancer、DeepVelo、VeloVI、STT(基于 ST)、SPATA(基于 ST)、TopoVelo(基于 ST)
⑥ HMRF(隐马尔可夫随机场):Startle(基于ST)
⑹ 分相
① 定义:将每个 RNA 转录物映射到父本或母本染色体,或至少映射到可区分的单倍型的过程。
表 10. 分相原则
> ② 主要在 F1 杂交小鼠模型中进行研究:RNA 转录物分别映射到每个单倍型或基于 SNP 的组合参考。
③ RNA-seq 定相类型:Lapels and Suspender pipeline、Eagle2、SHAPEIT、WhatsHap。
④ Hi-C 定相类型:HARP、HaploHiC、ASHIC、HiCHap(欧姆)。
⑺ ecDNA
① FISH检测:相当于实验方法。
② 短读长测序检测:Circle-Map、nf-core/circdna、ECCsplorer
④ Hi-C检测:EagleC、NeoLoopFinder。
⑻ HMM(隐马尔可夫模型)
① χ = {Xi} 是马尔可夫过程,Yi = phi(Xi) (其中 phi 是确定性函数),则 y = {Yi} 是隐马尔可夫模型。
② 鲍姆-韦尔奇算法
○ 目的:学习HMM参数
○ 输入:观察到的数据(例如 DNA 序列列表)
○ 输出:HMM的初始概率、状态转移概率、发射概率
○ 原理:EM(Expectation Maximization)算法的一种
○ 公式
○ Bk:状态 k 的初始概率
○ Akl: 从状态 k 到 l 的转换次数
○ Ek(b): 状态 k 的观测 b 的发射次数
③ 维特比算法 (ref)
○ 目的:给定 HMM 找到最可能的隐藏状态序列
○ 输入:HMM 参数和观测数据
○ N:可能的隐藏状态数
○ T:观测数据的长度
○ A:状态转移概率,akl = 从状态 k 转移到状态 l 的概率
○ E:发射概率,ek(x) = 在状态 k 下观察到 x 的概率
○ B: 初始状态概率
○ 输出:最可能的状态序列
○ 原理:利用动态规划计算最优路径
○ 步骤1. 初始化
○ bk: 状态 k 的初始概率,P(s0 = k)
○ ek(σ):在状态 k 下观测到第一个观测值 σ 的概率,P(x0 s0 = k)
○ 步骤 2. 递归
○ 计算每个时间步 i = 1, …, T 的先前状态的最大概率
»> ○ 计算反向指针 (ptr),存储最可能的先前状态
○ ptri(l) 用于存储最有可能转变到当前状态 l 的先前状态 k。
○ 步骤 3. 终止
○ 选择最后一个时间步的最高概率
○ 确定最优序列的最后状态
○ vk(i - 1): 状态 k 中前一时间步 i - 1 的最优概率
○ akl:从状态 k 转换到 l 的概率
○ 步骤 4. 回溯
○ 从 i = T, …, 1 开始通过 ptr 数组回溯,恢复最优路径
○ 示例
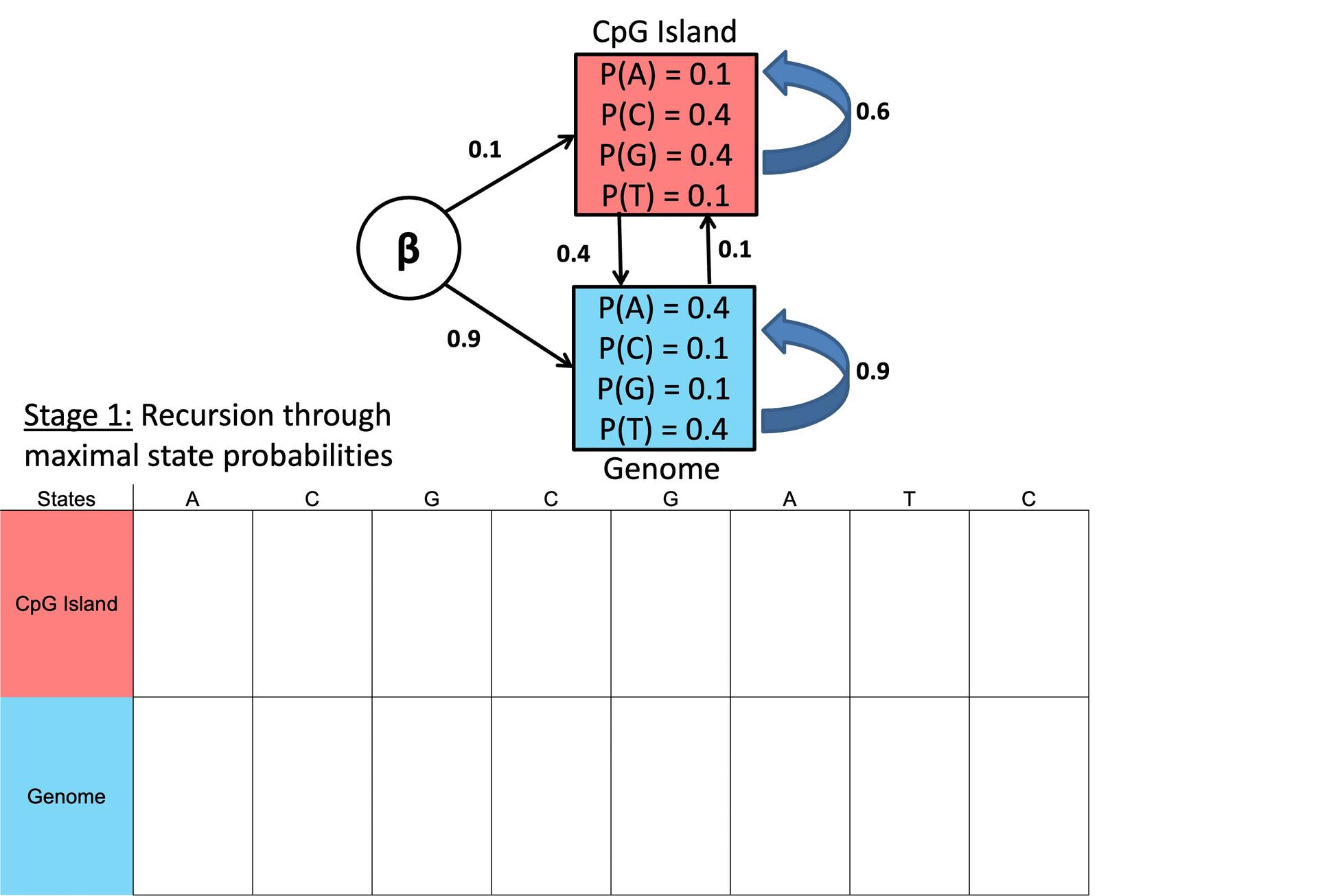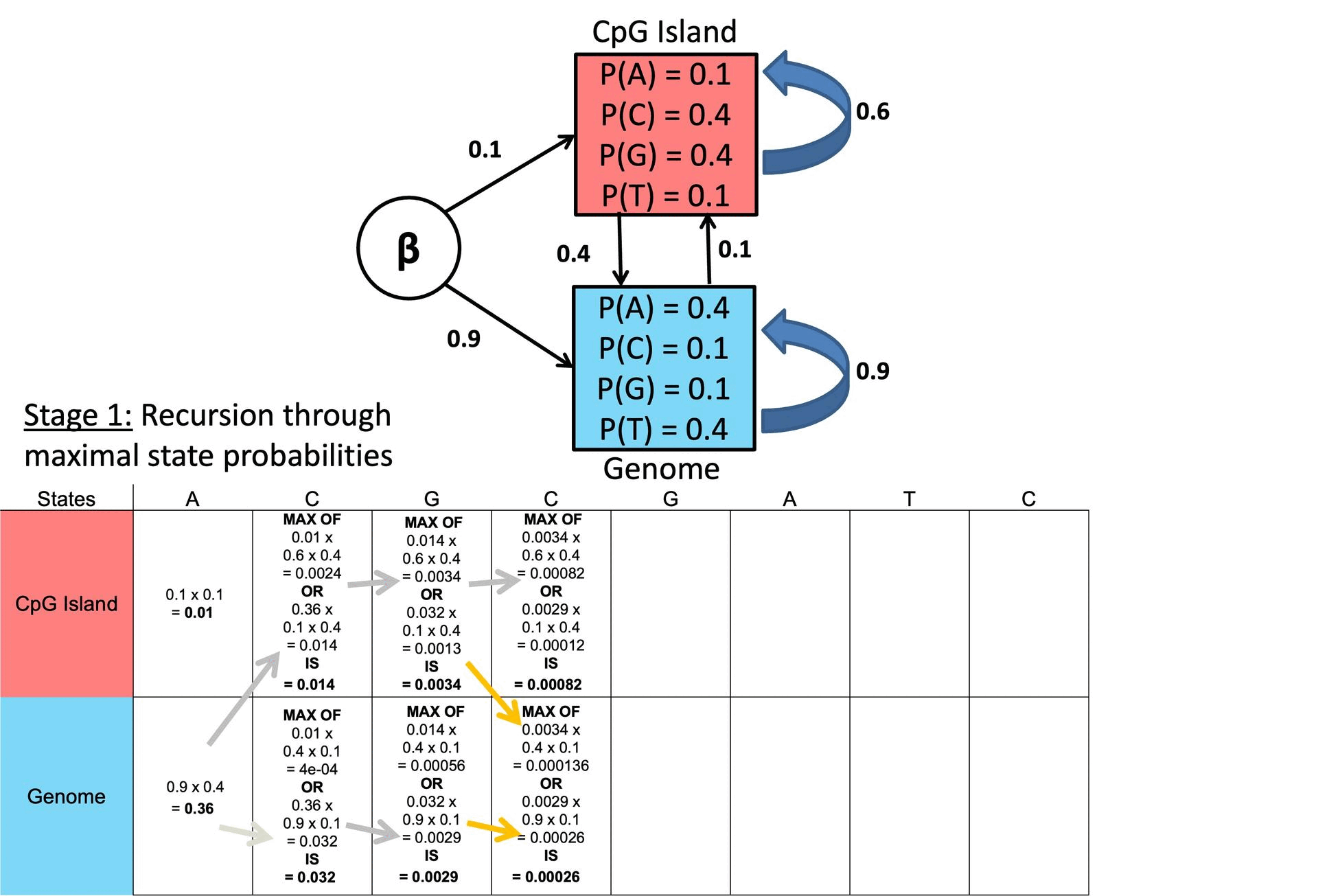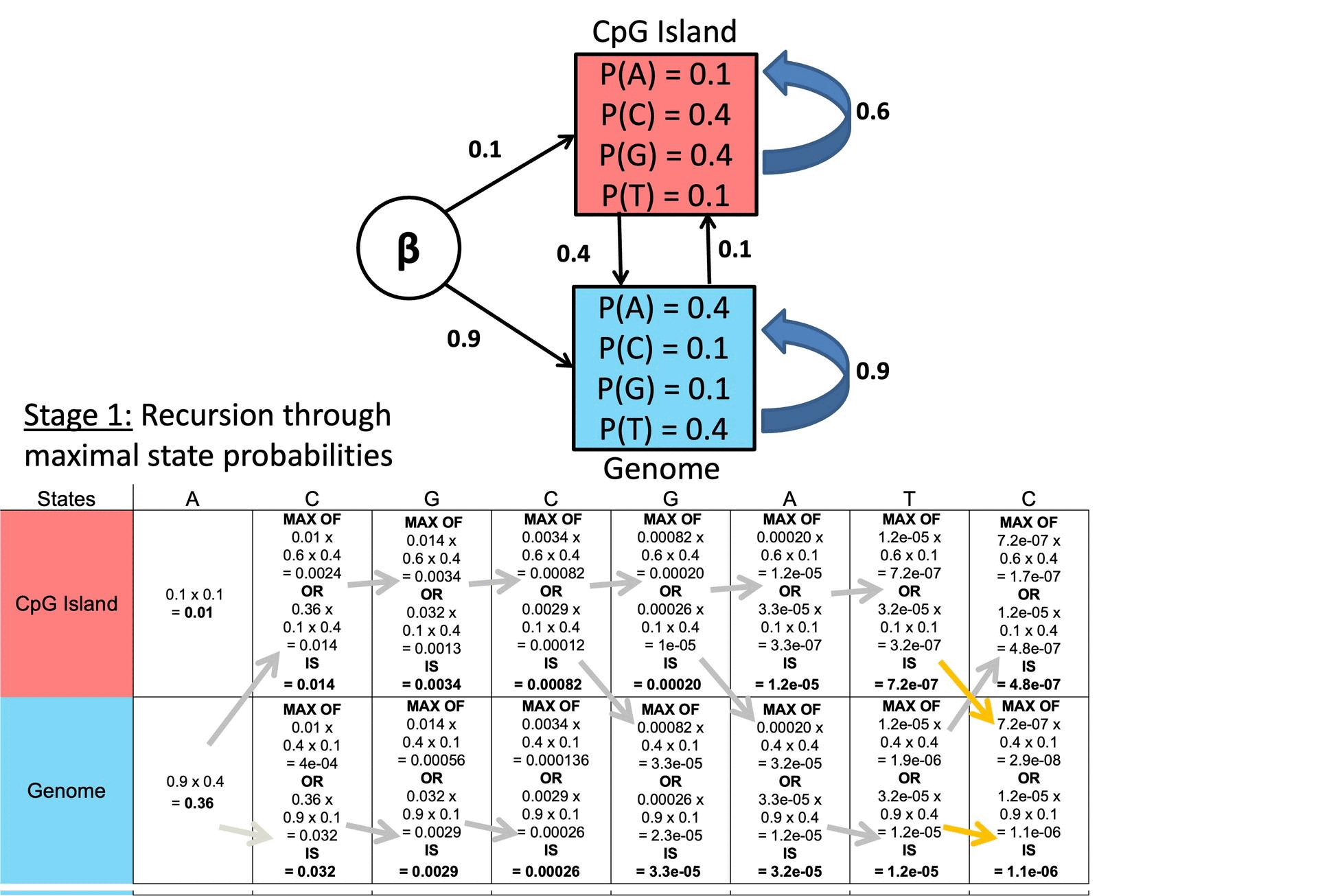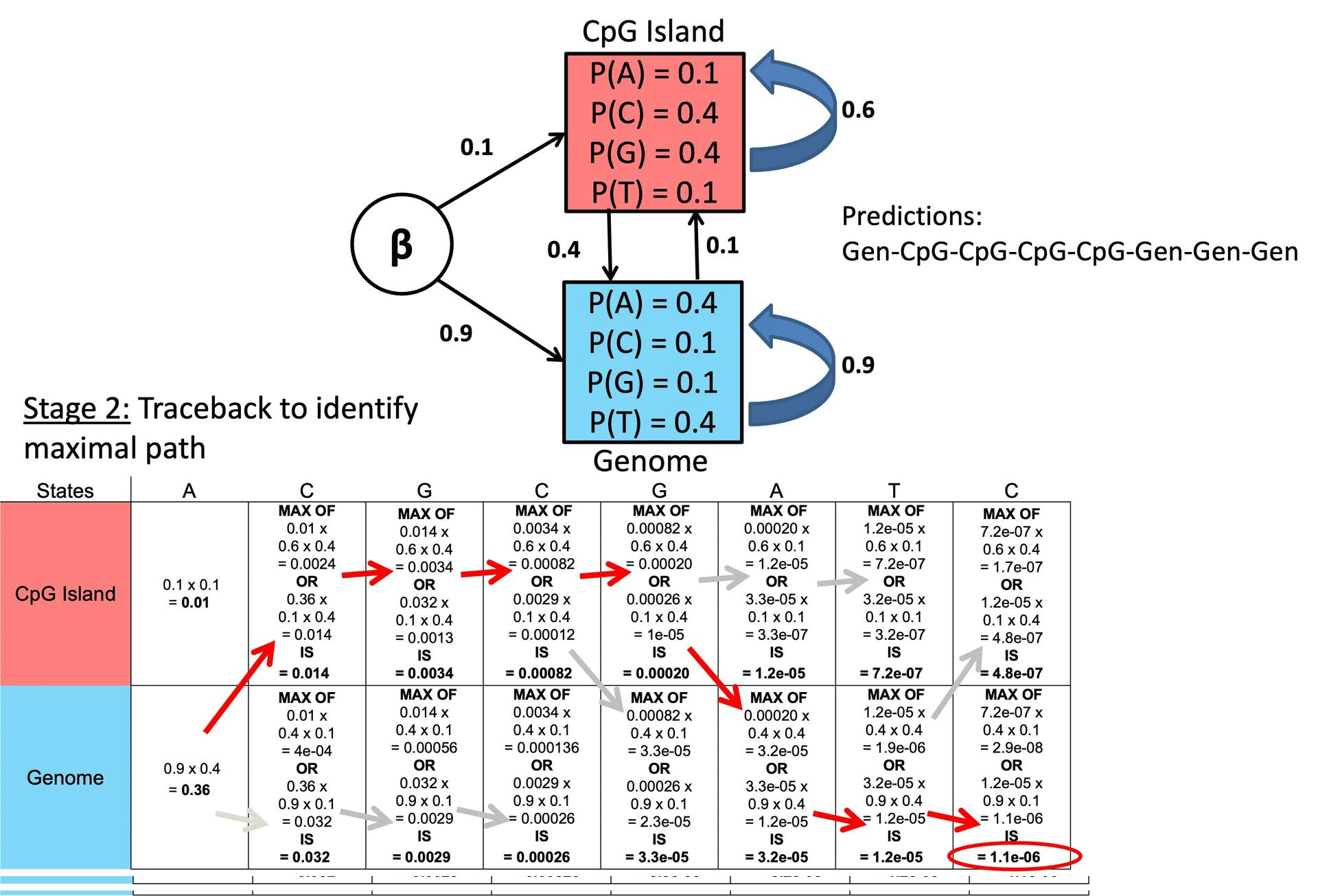
图 28. 维特比算法示例
○ Python 代码
受保护_35
④ 类型1. PSSM:更简单的HMM结构
⑤ 类型 2. Profile HMM:在以下方面优于 PSSM:
○ 轮廓HMM图
图 29. HMM 剖面图
○ M、I、D 分别代表匹配、插入、删除。
○ Mi 可以转换为 Mi+1、Ii 和 Di+1。
○ Ii 可以转换为 Mi+1、Ii 和 Di+1。
○ Di 可以转换为 Mi+1、Ii 和 Di+1。
○ 优点1. 能够对插入和删除进行建模
○ 优点 2. 转换仅限于有效状态遍历之间。
○ 优势 3. 州之间的界限得到更好的界定。
○ 示例 1. HMMER
○
hmmbuild:从一组比对序列创建轮廓 HMM。
○
hmmalign:将序列与配置文件 HMM 对齐。
○
hmmsearch:将配置文件 HMM 与序列数据库对齐。
○
hmmsca:根据配置文件 HMM 数据库比对序列。
○ 示例 2. HMMSTR
○ 示例3. SAM(序列比对和建模)
○ 示例 4. Pfam
○ 大量蛋白质家族的集合,每个家族都由多个序列比对和剖面 HMM 代表。
○ Pfam-A:一套高质量、手动策划的家庭配置文件。
○ Pfam-B:通过自动化方法 (ADDA) 识别的一组质量较低的家族。
14。 Advanced 3. 表观基因组学分析
⑴ 类型1. 【基因功能鉴定】(https://jb243.github.io/pages/1431#footnote_link_67_50)
① 测序类型
○ Perturb-seq:用不同的 gRNA 文库处理 Cas9 表达细胞,然后同时对 gRNA 和 mRNA 进行测序。
○ 体内扰动序列
② perturb-seq的DEG分析算法
○ CEDA
○ 玛吉克 RRA
○ MAGeCK MLE
○ G屏
○ 百吉饼2
○ 通用
③ 单细胞扰动预测
○ scGen:基于VAE
○ scPreGAN:基于GAN» ○ scVAEDer:VAE 和 DDN 的组合(最先进的扩散模型)
○ GeneFormer:基于LLM的基础模型
○ 注册会计师
○ CellOT
○ 齿轮
○ 生物王
○ PDGrapher
⑵ 类型2. 【转录调控识别】(https://jb243.github.io/pages/1431#footnote_link_67_51)
① 测序类型
○ BS-seq(亚硫酸氢盐测序):识别甲基化模式。
○ ChIP-seq(染色质免疫沉淀测序):识别转录因子的结合位点。
○ Hi-C(高通量染色质构象捕获测序):核染色质的3D折叠结构信息。
○ DNA 收报机磁带(Prime 编辑)
○ ENGRAM(增强子驱动的多重转录活性基因组记录)
○ ATAC-seq
○ NOMe-seq(核小体占据和甲基化测序)
○ MBD-seq
○ 核糖核酸测序
○ Bru-seq 和 BruChase-seq
② 峰值呼叫器(Peak finder)
○ 可用于 ChIP-seq、CUT&RUN、CUT&Tag、MeDIP-seq、MmethylCap-seq、hmeDIP-seq、DNase-Seq、ATAC-seq。
○ 步骤1. 读取比对和QC:ChIP-seq通常是单端测序,因此很简单。 CUT&Tag 分析使用双端测序。
○ 步骤 2. 移位大小估计(读取扩展):排列单端数据的 + 和 - 链读取(对于 ChIP-seq)。例如,当 + 链和 - 链上有两个相距一定距离的峰时,移动 1/2 片段大小会将它们合并为单个峰。
○ 步骤 3. 确定窗口大小:大多数寻峰器使用滑动窗口。
○ 步骤 4. 峰检测:识别 ChIP 样品中对照样品中没有的峰。涉及统计测试。
○ 步骤 5. 处理伪影:重复、链差异、链移位等。
○ 步骤 6. FDR 估计
○ 步骤 7. 处理重复样本,例如印尼盾。
○ 步骤 8. 下游分析,例如主题丰富。
⑶ 类型3. 【翻译后调控识别】(https://jb243.github.io/pages/1431#footnote_link_67_52)
① scRibo-seq
② STAMP-RBP
⑷ 类型4. 可编程单元功能
① 雷达
② LADL(光激活动态循环):光激活基因表达
15。 Advanced 4. 特殊转录组学分析
⑴ scRNA-seq 分析
① 归一化:SAMstrt (Katayama et al.)、BASiCS (Vallejos et al.)、GRM (Ding et al.)、Simple Norm (Satija et al.)、scran (Lun et al.)、SCnorm (Bacher et al.)、Linnorm (Yip et al.)、RUV-III-NB、sctransform、Giotto、SpaNorm
② 数据插补:读取插补、KNNimpute、scIGAN、MAGIC、VIPER、DeepImpute、SAUCIE
③批量效果去除:见上文。
④ 细胞周期估算:Cyclum、Cyclops、Oscape
⑤ 细胞类型预测:SINCERA、SC3、ACTINN、scVI、CSCORE(细胞类型特异性相关性)
⑥ CNV/亚克隆:见上文。
⑦ 系统发育树:SCARLET、Monovar
⑧ 细胞轨迹:见上文。
⑨ 基因-基因相互作用:PINNACLE、scNET、scLINE、GSPA
○ PINNACLE:结合了 scRNA-seq 和蛋白质-蛋白质相互作用 (PPI) 信息
○ scNET:结合了 scRNA-seq 和蛋白质-蛋白质相互作用 (PPI) 信息
○ 目标:了解更准确的表达模式和基因相互作用结构
○ 细胞间相似性 → 使用 KNN 图
○ 基因-基因关系 → 使用 PPI 网络
○ 内积解码器 → 恢复基因-基因关系
○ 全连接解码器 → 恢复基因表达值
○ scLINE:图嵌入方法
⑩ 细胞间通讯:见上文。
⑪基因网络:SCINET、SCENIC
○ 风景区»> ○ 识别调节子,定义为转录因子及其调节的基因
○ 它提供调节子特异性得分 (RSS),表明给定的调节子是特定于单个细胞类型还是在簇之间共享
⑫ 基因插补:scTransform、SAVER、MAGIC、DeepImpute
⑬ 单细胞扰动预测
○ scGen:基于VAE
○ scPreGAN:基于GAN
○ scVAEDer:VAE 和 DDN 的组合(最先进的扩散模型)
○ GeneFormer:基于LLM的基础模型
○ 注册会计师
○ CellOT
○ 齿轮
○ 生物王
○ PDGrapher
⑭ scRNA-seq/ST:DTSG
① 基于条形码(基于斑点)的转录因子:10x Visium 等。
② 基于图像(基于FISH)的转录因子:10x Xenium、Vizgen MERSCOPE、Nanostring CosMx 等。
○ Nanostring 与 10x Genomics 之间的专利纠纷 (‘23) ([ref1](https://www.businesswire.com/news/home/20230711643008/en/Delaware-District-Court-Permits-NanoString%E2%80%99s-Counterclaims-that-10x-Genomics-and-Harvard-Violated-the-Antitrust-Laws), ref2) → Nanostring 破产 (ref) 和收购([参考](https://www.businesswire.com/news/home/20240310548568/en/Patient-Square-Capital-a-Leading-Health-Care-Focused-Investment-Firm-Agrees-to-Acquire-NanoString-Technologies))
③ ST分析下游管道
○
Comprehensive Pipeline:Seurat、Squidpy、Scanpy、 [乔托](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02286-2),[空间数据](https://www.nature.com/articles/s41592-024-02212-x), ezSingleCell、SPACEc (谷歌Colab)、Sopa (Google Colab)
○ 降维和聚类:Seurat、Scanpy、Giotto、SPATA、STUtility» ○ 识别空间域:SPACEL、STAGATE、GraphST、 stLearn、RESEPT、Spatial-MGCN、 SpaGCN、ECNN、SEDR、 JSTA、STGNNks、 conST,CCST, BayesSpace、SpatialPCA、DRSC、Giotto-H、Giotto-HM、Giotto-KM、Giotto-LD、 Seurat-LV、Seurat-LVM、Seurat-SLM、IRIS、NeST、 SC-MEB、NovoSpaRc、UTAG、 BANKSY、NSF、BASS、SpatialLDA、 CeLEry、SpaGene、DeepST、 CellTrek、多层、RESEPT、Space-GM、SOTIP、SpaTM-S、scNiche、 Novae、MuCoST、STAIG、SPADE、HMRF、SpiceMix、 MENDER、SpaceFlow、细胞邻域聚类 (CNC)、GASTON、SpatialTopic、SpatialLDA、CytoCommunity(基于空间组学的 GNN)、 SpatialZoomer、CAST、ADEPT、ConGI、PRECAST、DR.SC、NicheCompass、BINARY
| 算法 | 所需空间信息 | 所需的组织学信息 | 所需簇数 | 程序语言 | |
|---|---|---|---|---|---|
| 贝叶斯空间 | 是的 | 没有 | 是的 | 右 | |
| DRSC | 可选 | 没有 | 可选 | 右 | |
| 乔托-H | 没有 | 没有 | 是的 | 右 | |
| 乔托-HM | 是的 | 没有 | 是的 | 右 | |
| 乔托-KM | 没有 | 没有 | 是的 | 右 | |
| 乔托-LD | 没有 | 没有 | 没有 | 右 | |
| 修拉-LV | 没有 | 没有 | 没有 | 右 | |
| 修拉-LVM | 没有 | 没有 | 没有 | 右 | |
| 修拉-SLM | 没有 | 没有 | 没有 | 右 | |
| 水疗细胞 | 没有 | 是的 | 是的 | 蟒蛇 | |
| SpaCell-G | 没有 | 没有 | 是的 | 蟒蛇 | |
| SpaCell-I | 没有 | 是的 | 是的 | 蟒蛇 | |
| 斯帕GCN | 是的 | 没有 | 可选 | 蟒蛇 | |
| SpaGCN+ | 是的 | 是的 | 可选 | 蟒蛇 | |
| 学习 | 是的 | 是的 | 没有 | 蟒蛇 |
表 11. 空间聚类方法
○ 贝叶斯空间
○ 采用贝叶斯方法。
○ 物理距离较近的空间位置权重较高。
○ 其性能可能受到马尔可夫随机场的固定平滑参数的限制。
○ 对于高通量空间转录组数据,计算上不可扩展。
○ 包括批量效应校正。
○ SC-MEB
○ 通过能够优化平滑参数的“经验贝叶斯方法”执行空间聚类。
○ 使用基于迭代条件模式的期望最大化方法来估计其参数,以提高其计算效率和高吞吐量数据的可扩展性。
○ novoSpaRc
○ 目标是尽量减少表达和空间数据中最短路径之间的差异。
○ 如果 2 个细胞在表达方面接近,则它们可能在空间上接近。
○ 它根据 scRNA-seq 数据的基于相关性的距离,从 KNN 图计算最短路径长度。
○ 内斯特
○ 步骤 1. 计算整个转录组中的单基因表达热点
○ 步骤2. 构建热点相似网络
○ 步骤 3. 网络中的社区被平均为表示共享表达模式的共表达热点
○ SEDR
○ 使用 GNN。
○ 阶梯
○ 使用 GNN。
○ 太空GM
○ 使用 GNN 执行监督学习。
○ 乌塔格
○ 无 GNN 的细胞注释
○ 包括批量效应校正。
○ 空间主成分分析»» ○ 使用概率 PCA 中的核矩阵来建模空间相关性。
○ 索蒂普
○ 基于单元邻域的网络对单元进行聚类。
○ 芹菜
○ 基于组织学和细胞结构的手动注释。
○ 空间主题
○ 使用细胞类型标签来识别空间域。
○ 识别空间可变基因(SVG):Moran’s I、Geary’s C、Ripley’s K、最近邻距离函数 G、景观形状索引、 SPARK (代码), SPARK-X (基于 R), SpatialDE(基于Python)(代码),SpaGCN, ST-Net、STAGATE、HisToGene、 CoSTA、CNNTL、SPADE、 DeepSpaCE、conST、 Spatial-MGCN、STGNNks、SpatialScope、 nnSVG(基于 R)、Hotspot、MERINGUE(基于 R)、 Trendsceek (代码), HMRF (代码), Celina、Crescendo、Giotto k-means(基于 R)、Giotto 秩(基于 R)、SomDE (基于Python)、Seurat、Scanpy、GPcounts、SpaTM-S、SpatialCorr、Sinfonia、标记变异函数(基于R的Seurat)、 SpaNormSVG、RayleighSelection、MULTILAYER、HRG、scGCO、SpaGene、C-SIDE、CTSV、BSP、DESpace、HEARTSVG、 BinSpect、Sepal、BOOST-GP、BOOST-MI、SMASH、SVGbit、PROST、BOOST-HMI、spVC、SpaGFT、SingleCellHayStack、Belayer、GSPA、 stIHC(层次聚类),SpaceExpress
图 30. SVG 检测方法的层次总结
○ 莫兰 I
○ –1 ~ 1:值越高表明正相关性和空间聚类性越强
○ 值小于 0 表示负空间自相关,而值大于 0 表示正空间自相关。
○ 基于 R 的 Seurat、基于 Python 的 Squidpy
○ Geary 的 C
○ 值越接近0表示正相关,意味着相邻点表现出相似的表达
○ 值大于 1 表示负空间自相关,而值小于 1 表示正空间自相关。
○ 基于Python的Squidpy
○ 空间DE»» ○ 高斯过程回归
○ 将表达变化分解为空间成分和非空间成分。
○ 根据位置之间的成对空间距离通过空间协方差建模的空间分量。
○ 非空间分量表述为噪声。
○ 计算复杂度低于 Trendsceek。计算复杂性仍然与空间位置的数量成立方关系。
○ 火花
○ 广义泊松回归
○ 具有多种核的生成模型:使用每个核计算 p 值,并利用柯西组合规则来组合 p 值。
○ 检测具有空间变异的基因。
○ 计算复杂度低于 Trendsceek。计算复杂性仍然与空间位置的数量成立方关系。
○ SPARK-X
○ 非参数法
○ 使用基因表达的协方差矩阵和空间坐标的协方差矩阵。
○ 如果基因表达与空间坐标无关,则两个协方差矩阵的乘积将会很小。
○ 计算复杂度低于 Trendsceek、SpatialDE 和 SPARK。
○ MERINGUE:空间自相关测量
○ nnSVG:最近邻高斯过程
○ 趋势科技
○ 标记点工艺原理
○ 基因表达(标记)
○ 空间位置(点)
○ 测试分数和分数分布的相关性。
○ 计算复杂度高
○ 斯帕GCN
○ GCN 方法
○ 整合基因表达数据、空间位置信息和组织学图像。
○ 聚合来自每个节点邻居的特征信息可以改善局部基因表达模式的识别,从而改善空间可变基因的识别。
○ 与 SPARK 和 SpatialDE 相比,SpaGCN 的计算速度更快,内存效率更高。
○ SVCA
○ SVCA 使用具有加性协方差的高斯过程来模拟基因表达的变化。
○ 分解为内在+环境+相互作用效应:通过整合基因表达和空间差异来建模相互作用。
○ 塞莉娜
○ 多内核策略
○ GSPA(基因信号模式分析)
○ 将过渡矩阵的乘积(=随机游走)应用于小波分析:原理类似于使用拉普拉斯矩阵应用傅立叶变换。
○ 它不像传统谱聚类那样使用拉普拉斯矩阵,而是使用从细胞-细胞图派生的马尔可夫转移矩阵 $P$ 的幂 Pt 来捕获不同扩散时间 $t$ 的信息。
○ 通过利用较短的扩散时间 $t$ 对局部模式敏感而较长的时间反映更多全局结构的特性。
○ 转移矩阵 P = AD-1:度数越高的节点在转移矩阵中的权重越低。
○ ψj = P2j-1 - P2j 对应于 Δt。» ○ 增强基因表达分辨率 (GER): BayesSpace, XFuse、DeepSpaCE、 HisToGene、SuperST、 TESLA、iStar、Thor(抗收缩马尔可夫图扩散方法)、TransformerST、Spotiphy(接受输入) scRNA-seq、H&E、ST)
○ iSTAR
○ 用于增强空间转录组学的分辨率。使用通过 DINO 方法训练的基于 BEiT 的模型。
图 31. iSTAR 数据准备图
○ 步骤 1. 将给定图像划分为 256 × 256 块。
○ 步骤 2. 将每个补丁细分为 16 × 16 子补丁。
○ 步骤3. 将ViT(表示为f2)应用于每个子补丁以获得384维向量。
○ 步骤4. 收集384维向量以创建16 × 16 × 384数据结构,然后应用另一个ViT(表示为f1)以获得192维向量。
○ 步骤 5. 收集 192 维向量并应用 ViT(表示为 f0)。
○ 特征和损失函数公式
○ 基因插补:LIGER、SpaGE、 stPlus、Seurat、七巧板、gimvI、 GeneDART、NovoSpaRc、spARC、BLEEP、SpaOTsc
○ 基因-基因相互作用:scHOT、GCNG、MISTy、MESSI、SEAGAL (Google Colab)、scMultiSim
○ 细胞间相互作用(CCI):Giotto、MISTy、stLearn、 [GCNG](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02214-w),[conST](https://www.biorxiv.org/content/10.1101/2022.01.14.476408.full), COMMOT、NCEM、七巧板、 对话、CellTrek、SVCA、spata2、CellPhoneDB v3、SCOTIA、STOPover、 cytosignal、SpatialDM、SpaTalk、stMLnet、HoloNet、DeepLinc、scMultiSim» ○ 细胞类型解卷积/识别:MIA、Sterescope、 RCTD、Cell2location、DestVI、 STdeconvolve、SPOTlight、 SpatialDWLS、GIST、 GraphST、DSTG、七巧板、 CellDART、spSeudoMap、TACCO、 平滑、CARD、 Cellscope、Starfysh、Seurat、AdRoit、Spatial-ID(来自 scRNA-seq 的注释转移)、 SpaTM-G、SpaOTsc、NLSDeconv、Redeconve、SpaCET、CellTrek、SpatialPrompt、SPADE、SpatialDecon、SONAR、AntiSplodge、UniCell Deconvolve、SpatialDDLS、TransformerST、 DEEPsc、RedeHist、LETSmix、scResolve、SPACEL-Spoint、STEM、SpaDecon、Bulk2space、SpatialcoGCN、SD2、GNNDeconvolver、GTAD、STdGCN、STRIDE、SMART、stVAE、POLARIS、STIE、MUSTANG、BayesTME、RETROFIT、SpatialScope、ST-assign、MAST-Decon、EnDecon、 Cottrazm、CellsFromSpace、FAST、CellPie、SpiceMix、LANTSA、NovoSparc、scDOT、DOT、SpaTrio、CytoSPACE、LANSTA、BayesPrism、BASS、SDDLS、BANKSY、 蛋白酥皮、SpaGCN、BayesSpace、FICT、 CMAP、SpatialZoomer、Spotiphy(采用 scRNA-seq 和 ST 的输入)
图31. 细胞类型反卷积算法的类型
» ○ 细胞分割:Watershed、Otsu、Cellpose、JSTA、 Baysor、GeneSegNet、 SSAM、ClusterMap、SCS、 QuST、Proseg、Bioturing 分割 (github)、 InstanSeg、StarDist、CellViT (github、 应用), HistAI (github), BIDCell, 图,Comseg, Points2Regions,Sainsc, UCS、HoVer-Net、Triple U-Net、CDNet、Omnipose、CPP-Net、PathoSAM、NuLite、ilastik、MESMER
○ Cellpose:训练人工神经网络以确定像素的梯度是否指向细胞内部。生成
.tif和.npy格式的核和细胞分割结果。
○ QuST:QuPath 的扩展。生成 GeoJSON 格式的核和细胞分割结果。
○ Baysor:使用贝叶斯混合模型。考虑转录本位置·组成、细胞大小·形状来确定细胞边界。
○ Proseg:考虑转录本位置·组成、细胞大小·形状来确定细胞边界。使用细胞 Potts 模型、MCMC 和转录物重新定位。
○ BIDCell:自监督深度学习
○ Points2Regions:无监督聚类、K-means聚类
○ 无分割方法:FICTURE、Points2Regions、 ComSeg
○ 空间利基:NicheNet、Nicheformer、CellCharter (Google Colab)、 GraphSAGE、SIMVI、NicheDE、COVET、SpaTopic、SpatialLDA
○ Cellcharter:采用 scVI 嵌入和 GMM 聚类。
○ SpaTopic:使用细胞类型和空间域作为输入导出与微环境相对应的主题。
○ ST 细胞扰动:Vespucci、River、Perturb-STNet、stFormer、Morpheus、CellAgentChat、CONCERT
○ 图像对齐(图像配准):DiPY、bUnwarpJ (ImageJ)、STalign、SpatialSPM» ○ 图像到基因表达:NSL、DeepSpaCE、 ST-Net、HisToGene、 Hist2ST、THItoGene、 TCGN、DeepPT、BLEEP、GHIST、GeneCodeR、iSTAR、ssClassify
○ 染色归一化:Macenko 归一化
○ 3D 重建:PASTE、SPACEL、STAGATE、STUtility(仅使用组织学图像,不使用空间基因表达)、Splotch(适用于较旧的 ST 平台;需要手册)注释)、GPSA、XVFI、InterpolAI、spatio
○ 粘贴
○ 一种通过考虑基因表达相似性和空间距离来合并和对齐来自同一组织的多个 ST 切片的算法。
○ 使用 NMF 提取基因表达的潜在特征,并通过最优传输定理将潜在特征和空间距离结合起来。
○ 能够跨多个切片进行更准确的聚类。
○ 模式:成对切片对齐、中心切片积分。
○ 不使用组织图像。
○ 每个点的基因表达值使用现有的原始计数或归一化计数,但该点的位置在 3D 空间中进行调整。
○ STutility
○ 仅使用组织图像,不考虑基因表达或空间坐标。
○ 斑点
○ 针对较旧的 ST 平台数据而开发。
○ 需要手动注释。
○ XVFI:3D生物医学图像重建
○ 国际刑警组织
○ 用于恢复 3D 生物医学图像堆栈中丢失或损坏图像的模型(例如 H&E、MRI 或荧光图像)。
○ FILM(大运动帧插值)模型的修改版本,最初用于视频帧插值。
○ 最简单的情况是,人们可能会考虑线性插值,但这种模型明显更准确。
○ 不仅能够在 2D 图像之间进行插值,还能够对插值后的 2D 图像进行 3D 渲染。
○ CNV 推理:SpatialInferCNV、SPATA2 ([演示](https://drive.google.com/file/d/1BHO1-zHNDPfbEmL-2AkHH7krdmEG-VfE/view?usp=drive_link)),[STmut](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03121-6), STARCH、CalicoST
○ 轨迹分析:stLearn、SpaceFlow、SPATA2、 Startle、Spateo、MOSCOT、SLAT、Hotspot、cell2fate、GASTON、STT、TopoVelo、STORIES(使用最佳传输)、PRESCIENT
○ 基因panel设计:scGIST、scGeneFit、geneBasis、SMaSH、RankCorr、PERSIST、gpsFISH
○ 空间数据模拟:scDesign3
图 32. ST 分析下游管道
> ④ 3D空间转录组学(ref1, [参考2](https://www.nature.com/articles/s41586-023-06808-9),[参考3](https://www.biorxiv.org/content/10.1101/2023.07.21.550124v1),[参考4](https://www.science.org/doi/10.1126/science.adn9947), 参考5)
⑤ 亚细胞 ST:最近邻、InSTAnT、 TopACT(细胞类型分类)、基于 APEX-seq 的图谱、Bento、 TEMPOmap,亚细胞 mRNA 动力学建模,[Rustem 等等人](https://www.biorxiv.org/content/10.1101/2024.11.27.625536v1),[鹏飞等人](https://www.biorxiv.org/content/10.1101/2024.12.23.630033v1.full),CellSP, SPRAWL (Bierman et al. 2024)
⑶ 时间转录因子分析
① 类型1. 短时间序列数据
○ STEM(短时间序列表达矿工)
○ 时代矢量
② 类型2. 长时间序列数据:类似于一般实验组的分析
③ 类型 3. 时间排序
○ 记录序列
○ 实时测序
○ TMI
○ 分子记录
⑷ 时空组学
① 轨道 (单分子DNA折纸旋转测量)
② 4D时空MRI或超极化MR
③ in vivo 4D 组学透明小鼠
⑸ 【空间三组学】(https://www.biorxiv.org/content/10.1101/2024.07.28.605493v1)
① DBiT ARP-seq(ATAC + RNA + 蛋白质)
② DBiT CTRP-seq (CUT&Tag (H3K27me3) + RNA + Protein)
16。 Advanced 5. 数据库利用率
⑴ 生物信息学资源
① 示例:PubMed、NCBI、bioRxiv、BioStars、Bioinformatics Stack Exchange、Stack Overflow
② 数据存储库
○ ArrayExpress (EBI)
○ 基因表达综合 (GEO):不再支持 fastq-dump。
○ 基因组RNAi、dbGAP
○ 欧洲基因组-现象档案馆 (EGA)
○ 相互作用蛋白数据库(DIP)
○ 完整
○ 日本基因型-表型档案馆 (JGA)
○ NCBI PubChem 生物测定
○ 基因组表达档案 (GEA)
○ GWAS 目录
○ UCSC 基因组浏览器
③【网页抓取】(https://jb243.github.io/pages/2291)
⑵ 小分子数据库
① 综合小分子数据库:以矢量格式提供约800,000个小分子生理活性数据的数据库
② 【AlphaFold2数据库】(https://alphafold.ebi.ac.uk/):拥有2亿个蛋白质结构数据的数据库
③ ensembl:转录组数据库
⑥ SGC(化学探针):提供独特的探针集合以及相关数据、对照化合物和使用建议
⑶ 空间转录组数据库
① HCA
② HuBMAP
③ SODB
⑥ 翱翔
⑦ HTAN
⑧ 【艾伦大脑图谱】(https://portal.brain-map.org/)
⑨ CZ CELLxGENE
⑩ 单细胞门户
⑪ 人类细胞图谱数据门户
⑫Bgee
⑬ 地理
⑷ 药物基因组数据库
① NCBI dbSNP
② 侏儒AD
③ 医药变量
④ 医药知识库
⑤ NCBI PubChem
⑥ 布罗德研究所CMAP
⑦ CTD
⑧ 康普托克斯
⑨ 药物库
⑩ Stitch(化学物质相互作用搜索工具)
⑪ ToppFun
⑫ DepMap:提供相应细胞系的表达数据和谱系信息。
⑬ L1000CDS2
⑭ L1000FWD
⑮ GDSC(癌症药物敏感性基因组)
⑯CCLE
⑰ ClinicalTrials.gov:提供每种药物的临床试验进展信息
⑱Cortellis:提供每种药物的临床试验进展信息。
⑲ 抗体协会:提供抗体临床试验进展信息。
⑳ PRISM:提供数百种癌细胞系的大规模药物反应数据。
⑸ 临床和非临床数据库
① TCGA(癌症基因组图谱)
○ 基于 DNA、RNA、蛋白质表达和表观遗传因素的多种人类肿瘤分析数据
○ 【如何获取TCGA数据】(https://jb243.github.io/pages/1694)
② GWAS目录
○ 提供有关基因组相关信息的精选信息和教育资源,以识别因果变异并了解新疗法的疾病机制
③ MGI(小鼠基因组信息学)
○ 收集小鼠突变、表型和疾病数据的数据库。每个基因本体(GO)都组织良好
④ 开放目标平台
○ 表型数据的集成平台,例如与某些疾病相关的特定目标相关的表达、共定位和优先级签名
⑤ 英国生物样本库
○ 代谢组:样本120,000。根据 2006 年至 2010 年采集的血液进行测量
○ 血液生物标志物:500,000 个样本。根据 2006 年至 2010 年采集的血液进行测量
○ 基因组(GWAS、WES、WGS):样本 500,000。根据 2006 年至 2010 年采集的血液进行测量
○ 摘要级临床记录:样本 500,000 份。医院诊断(ICD 代码)和首次诊断日期
○ 记录级临床记录:样本25万。特定日期的诊断/处方记录。跟踪的起始年份如下
○ 1997 代表英格兰
○ 1998 年 威尔士
○ 1981 苏格兰
⑥ 侏儒AD
○ 聚合人类基因组变异数据的大型公共数据库:主要作为遗传变异和罕见疾病研究的参考。
○ 方法 1. Google Cloud:使用命令
$ gsutil ls gs://gcp-public-data--gnomad/release/。也可作为 BigQuery 数据集使用。
○ 方法 2. AWS:使用命令
$ aws s3 ls s3://gnomad-public-us-east-1/release/。使用 AWS 命令行界面。» ○ 方法 3. Azure:使用命令$ azcopy ls https://datasetgnomad.blob.core.windows.net/dataset/。可以通过 AzCopy 或 Azure 存储资源管理器进行访问。
○ 方法 4. 冰雹
○ 方法5. Terra
⑦ 其他临床数据库
| 国家 | 机构 | 临床数据 | 基因组数据 | 转录组数据 | 蛋白质组数据 | 成像数据 |
|---|---|---|---|---|---|---|
| 美国 | 已涂底 | 哦 | 哦 | |||
| 美国 | 美国全境 | 哦 | 哦 | |||
| 美国 | 国家糖尿病、消化和肾脏疾病研究所 | 哦 | 哦 | |||
| 美国 | 美洲基因组医学 Slim 倡议 | 哦 | 哦 | |||
| 英国 | 英国生物银行 | 哦 | 哦 | 哦 | 哦 | 哦 |
| 中国 | 嘉道理生物银行 | 哦 | 哦 | 哦 | 哦 |
表 12. 其他临床数据库
⑹ 政策数据库
① EQIPD质量体系:由8个国家和29个机构组成的欧洲临床前数据质量(EQIPD)联盟开发的新的非临床研究质量体系,适用于公共和私营部门
② FAIRsharing:有关数据库和数据政策相关数据和元数据标准的精选信息和教育资源
⑺ 网络平台
① Chemicalprobes.org:接收有关在药物研究和药物开发中寻找和使用化学探针的专家建议的门户网站
② European Lead Factory:创新药物开发的公私合作伙伴网站
③【基因型-组织表达项目】(https://www.gtexportal.org/home/):组织特异性基因表达与调控的公共资源项目
④ GOT-IT专家平台 : 促进学术研究人员和行业专家之间交流的平台,促进新的学术界与工业界的合作
⑤ SPARK 全球倡议:专注于交流专业知识和解决当前未满足的医疗需求、加强和开发项目的国际网络
输入:2021.10.02 13:49
修改:2023.07.11 11:19